![]()
1
БИОЛОГИЧЕСКОЕ ОКИСЛЕНИЕ.
ПОНЯТИЕ О МЕТАБОЛИЗМЕ.
Метаболизм – это совокупность химических реакций, протекающих в организме. При этом процессы, происходящие в просвете желудочно-кишечного тракта, не входят в понятие метаболизма, поскольку полость желудочно-кишечного тракта рассматривается как часть внешней среды. Метаболизм включает в себя более чем 100 000 разнообразных реакций, но существуют основные метаболические пути, построенные по единому плану. Такие пути могут быть линейными и разветвленными. Ферменты, катализирующие реакции, протекающие на этих путях, в организме объединены в мультиферментные системы. В мультиферментных системах продукт предыдущей реакции является субстратом для последующей.
Метаболизм – это двуединый процесс, складывающийся из 2-х частей: катаболизма и анаболизма. В ходе катаболизма происходит разрушение, расщепление сложных веществ до более простых. В процессе анаболизма организм синтезирует собственные сложные органические вещества из простых. Оба процесса связаны между собой большим числом реакций, хотя в клетке часто бывают пространственно разделены.
Однако, существуют химические реакции из числа обратимых, которые в равной степени можно отнести как к катаболизму, так и анаболизму. Принадлежность той или иной реакции к одному из этих процессов определяется тем, в какую сторону сдвинуто ее равновесие в данный момент времени.
СХЕМА ЭТАПОВ КАТАБОЛИЗМА
1-й этап. Образование мономеров из полимеров.
|
Полимеры |
———>Мономеры |
|
Белки ———— |
> Аминокислоты |
|
Крахмал ——— |
>глюкоза |
|
Жиры ———— |
>глицерин + жирные кислоты |
2-й этап. Превращение мономеров в ПВК и Ацетил-КоА.
3-й этап. Превращение Ацетил-КоА в конечные продукты катаболизма: СО2 и Н2О.
Для всех классов веществ последний этап катаболизма одинаков: на 3-м этапе образуется большинство субстратов митохондриального окисления — 4 вещества из 9 основных и 5-й субстрат — ПВК.
БИОЛОГИЧЕСКОЕ ОКИСЛЕНИЕ — это совокупность окислительных процессов в живом организме, протекающих с обязательным участием кислорода. Синоним — ТКАНЕВОЕ ДЫХАНИЕ. Окисление одного вещества невозможно без восстановления другого вещества. Окислительновосстановительных процессов в живой природе очень много. Часть окислительновосстановительных процессов, протекающих с участием кислорода, относится к биологическому окислению.
ИСТОРИЯ РАЗВИТИЯ УЧЕНИЯ О БИООКИСЛЕНИИ.
А. Лавуазье в конце XVIII века показал, что животный организм потребляет из воздуха кислород и выделяет углекислый газ. Сделал вывод, что горение и окисление — это одно и то же, что биологическое окисление представляет собой «медленное горение», происходящее в присутствии воды
ипри низкой температуре.
Вконце XIX века русские исследователи А.Н. Бах и В.И.Палладин, работая независимо друг от друга, предложили 2 основные теории для объяснения процессов, протекающих в ходе биологического окисления.
1-я теория: А.Н.Бах (1857-1946) полагал, что в живых клетках существуют особые ферменты — «оксигеназы», которые взаимодействуют с кислородом, образуя перекиси. Сам кислород является не очень активным окислителем. Зато перекиси («активный кислород») являются очень сильными окислителями и способны передавать кислород окисляемому веществу.
Эта теория известна как «перекисная» или «теория активации кислорода».
2
2-я теория: В.И. Палладин (1859-1922) создал теорию «активации водорода». Считал, что универсальным путем окисления является отнятие от веществ (субстратов) водорода с участием специальных ферментов — хромогенов. После этого водород, по Палладину, может передаваться или на молекулу кислорода с образованием воды, или на другие молекулы, восстанавливая их.
Впоследствии теория В.И.Палладина блестяще подтвердилась для процессов митохондриального окисления, а ферменты, принимающие непосредственное участие в отнятии водорода от субстратов, в настоящее время называются дегидрогеназами.
СОВРЕМЕННАЯ ТЕОРИЯ БИООКИСЛЕНИЯ
Согласно СОВРЕМЕННОЙ ТЕОРИИ БИООКИСЛЕНИЯ в нашем организме окисление может происходить двумя способами:
1. Путем отнятия водорода от окисляемого субстрата: сюда относятся МИТОХОНДРИАЛЬНОЕ ОКИСЛЕНИЕ и ВНЕМИТОХОНДРИАЛЬНОЕ ОКИСЛЕНИЕ ОКСИДАЗНОГО ТИПА.
2. Путем присоединения кислорода к окисляемому субстрату — так происходит внемитохондриальное ОКИСЛЕНИЕ ОКСИГЕНАЗНОГО ТИПА (старое название — МИКРОСОМАЛЬНОЕ окисление).
МИТОХОНДРИАЛЬНОЕ ОКИСЛЕНИЕ (МтО).
Система митохондриального окисления — мультиферментная система, постепенно транспортирующая протоны и электроны на кислород с образованием молекулы воды.
Все ферменты митохондриального окисления встроены во внутреннюю мембрану митохондрий. Только первый переносчик протонов и электронов — никотинамидная дегидрогеназа расположена в матриксе митохондрии. Этот фермент отнимает водород от субстрата и передает его следующему переносчику. Полный комплекс таких ферментов образует «дыхательный ансамбль» («дыхательную цепь»), в пределах которого атомы водорода отнимаются от субстрата, затем передаются последовательно от одного переносчика к другому, и, наконец, передаются на кислород воздуха с образованием воды.
Существует строгая последовательность работы каждого звена в цепочке переносчиков. Эта последовательность определяется величиной РЕДОКС-ПОТЕНЦИАЛА (ОКИСЛИТЕЛЬНО-
ВОССТАНОВИТЕЛЬНОГО ПОТЕНЦИАЛА, сокращенно — ОВП) каждого звена. ОВП — это химическая характеристика способности вещества принимать и удерживать электроны. Выражается в вольтах (V). Вещества с положительным ОВП окисляют водород (отнимают от него электроны), вещества с отрицательным ОВП окисляются самим водородом. Самый низкий ОВП имеет начальное звено цепи, самый высокий — у кислорода, расположенного в конце цепочки переносчиков. Таким образом, передача водорода идет от более низкого к более высокому ОВП. Перенос водорода и электронов возможен только в одном направлении — в порядке возрастания их ОВП: от -0.32V у никотинамидных дегидрогеназ (первого компонента главной цепи МтО) до 0.82V у О2, обладающего самым высоким редокс-потенциалом.
На одной из стадий происходит разделение атомов водорода на Н+ и электроны. Протоны остаются временно в окружающей среде, а электроны идут дальше по цепи и в ее конце используются для активации О2. Кислород является конечным акцептором электронов.
O2 + 4e ——> 2O-2 (полное восстановление кислорода)
Все реакции, происходящие в дыхательной цепи, сопряжены. Переносчики водорода и электронов расположены в строгом порядке, в соответствии с величиной их редокс-потенциала.
В настоящее время различают три варианта дыхательных цепей:
1)ГЛАВНАЯ (ПОЛНАЯ) ЦЕПЬ
2)УКОРОЧЕННАЯ (СОКРАЩЕННАЯ) ЦЕПЬ
3)МАКСИМАЛЬНО УКОРОЧЕННАЯ (МАКСИМАЛЬНО СОКРАЩЕННАЯ) ЦЕПЬ.
Сначала разберем их строение на примере главной дыхательной цепи.
3
I.ГЛАВНАЯ ДЫХАТЕЛЬНАЯ ЦЕПЬ
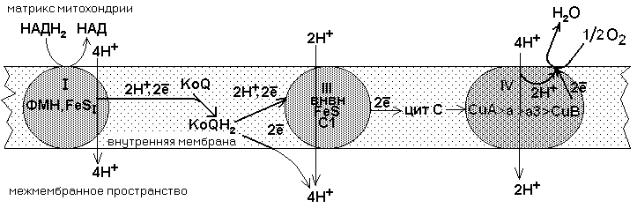
Главная дыхательная цепь — это три мультиферментных комплекса, встроенных во внутреннюю мембрану митохондрии. Обозначаются они латинскими цифрами – I, III и IV.
СХЕМА ГЛАВНОЙ (ПОЛНОЙ) ДЫХАТЕЛЬНОЙ ЦЕПИ МИТОХОНДРИАЛЬНОГО ОКИСЛЕНИЯ
Комплекс I – НАДН-KoQ-редуктаза, комплекс III – KoQH2-редуктаза, комплекс IV – цитохромоксидаза. Есть еще комплекс II – сукцинат-KoQ-редуктаза, но он существует отдельно от остальных комплексов и не входит в состав главной цепи.
Эти комплексы транспортируют водород от никотинамидных дегидрогеназ на кислород воздуха, в результате чего создается электрохимический градиент концентраций протонов — +. Он возникает на внутренней мембране митохондрий между матриксом и межмембранным пространством. Его составляют два основных фактора:
1)Электрический мембранный потенциал .
2)Градиент pH (осмотический или химический градиент).
+= — p
+ — положительная величина. Его можно выразить как в вольтах (V), так и в единицах энергии (кДж/моль). Изменение значения pH на одну единицу соответствует 0,06V или 5,7 кДж/моль.
Энергия + используется для следующих процессов:
1)Синтез АТФ.
2)Получение тепла (особенно важно для бурого жира и для мышечной ткани птиц).
3)Выполнение осмотической работы (транспорт фосфата в матрикс митохондрии).
4)Мышечная работа (в некоторых случаях).
Для человека наиболее важен синтез АТФ.
В полной цепи при окислении субстрата два атома водорода переносятся на НАД – кофермент никотинамидных дегидрогеназ.
Как видно из приведенной схемы, в полной цепи при передаче двух атомов водорода на кислород воздуха, в межмембранном пространстве оказываются 10 протонов, перенесенных сюда из матрикса.
Все переносчики встроены во внутреннюю мембрану митохондрий, кроме никотинамидных дегидрогенказ. Они составляют дыхательный ансамбль, тысячи таких ансамблей существуют в митохондрии и потребляют 90-95% кислорода, который используется клеткой. Два атома водорода отнимаются от субстрата и передаются на О2 с образованием Н2О. Разность потенциалов на двух концах полной цепи составляет 1.14V.
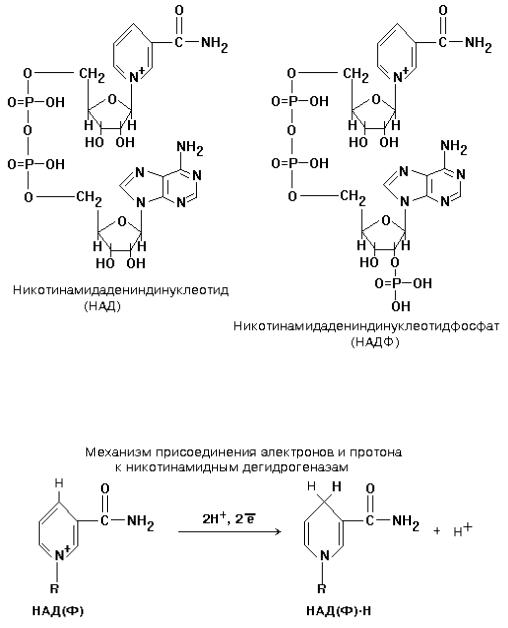
НИКОТИНАМИДНЫЕ ДЕГИДРОГЕНАЗЫ (НАДГ)
Небелковая часть этих ферментов представляет собой динуклеотид: НИКОТИНАМИДАДЕНИНДИНУКЛЕОТИД (НАД+) или НИКОТИНАМИДАДЕНИНДИНУКЛЕОТИДФОСФАТ (НАДФ+).
4
Студенты обязаны знать формулу НАД(Ф) и механизм присоединения к нему водорода. НАД(Ф) содержит производное витамина РР — никотинамид. (см. раздел «Витамины»).
НАД+ и НАДФ+ входят в состав каталитического центра НАДГ. Они являются КОФЕРМЕНТАМИ, так как связаны с белковой частью слабыми типами связей — могут легко диссоциировать. Они присоединяются к белковой части только в момент протекания реакции. Реакция, которую катализируют НАДГ — это реакция окисления субстрата.
Известно около 150 НАДГ, которые различаются по строению белковой части (апофермента). Апоферменты большей части НАДГ способны присоединять или только НАД, или только НАДФ, и лишь немногие способны соединяться и с тем, и с другим коферментами. НАДГ, участвующие в
митохондриальном окислении, находятся в матриксе митохондрий, в отличие от большинства других участников дыхательной цепи, которые встроены во внутреннюю мембрану. НАДГ можно встретить и в цитоплазме клеток. Мембрана митохондрий непроницаема для НАД(Ф), поэтому митохондриальный и цитоплазматический НАД(Ф) никогда не смешиваются. В митохондриях содержится очень много НАД и почти нет НАДФ, а в цитоплазме — наоборот — очень много НАДФ и почти нет НАД.
Из матрикса митохондриальный НАД Н2 отдает два атома водорода на «комплекс I», встроенный во внутреннюю мембрану митохондрий.
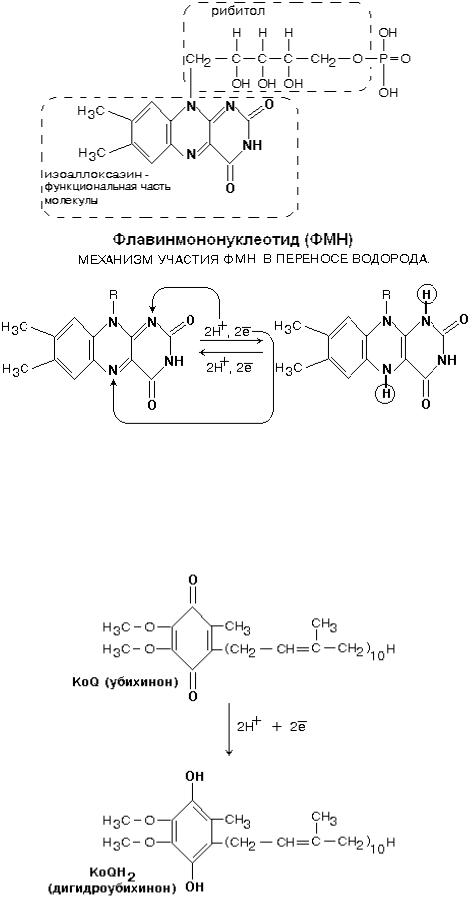
КОМПЛЕКС I
Всоставе комплекса находится 26 полипептидных цепей общей массой 800 кДа. Комплекс содержит следующие небелковые компоненты: Флавинмононуклеотид (ФМН), 5 центров FeS (железо-
серные центры): FeS1a, FeS1b FeS2, FeS3, FeS4.
Втранспорте водорода по дыхательной цепи в этом комплексе принимает участие ФМН.
5
Одновременно с протонами транспортируются и электроны. Наибольшие перепады редокспотенциала наблюдаются между железо-серными белками, расположенными в следующем порядке:
ФМН FeS1a FeS1b FeS3 FeS4 FeS2
Комплекс I – интегральный белковый комплекс. Используя энергию, выделяющуюся при переносе электронов по дыхательной цепи, он транспортирует 4 протона из матрикса в межмембранное пространство – комплекс I работает как протонный генератор. Точный механизм этого транспорта до сих пор неизвестен.
Далее комплекс I восстанавливает промежуточный переносчик KoQ (убихинон).
Это жирорастворимое низкомолекулярное вещество, содержащее длинную изопреновую цепь, не имеет белковой части. КоQ принимает водород от комплекса I. Образовавшийся КоQH2 отдает водород на комплекс III.
6
КОМПЛЕКС III.
В своем составе содержит цитохромы – сложные белки, содержащие небелковый компонент — простетическую группу, сходню по строению с небелковой частью гемоглобина – гемом.
1) Цитохромы b, имеющие в своем составе два типа простетических групп тетрапиррольной структуры — «гем». Известно два гема цитохромов: be, обладающий низким окислительновосстановительным потенциалом и bh с высоким окислительно-восстановительным потенциалом. Строение простетической группы цитохромов группы b, похожей на гем белка гемоглобина, представлено на рисунке. Его необходимо выучить.
2)FeSIII – железо-серный кластер.
3)Цитохром С1. Имеет в своем составе особый гем типа «с».
Друг от друга цитохромы могут отличаться:
|
1) |
Строением белковой части; |
|
|
2) |
Значением |
окислительно- |
|
восстановительного потенциала; |
||
|
3) |
Строением радикалов, расположенных по |
|
|
периферии гема; |
||
|
4) |
Присоединением гема к белковой части – в |
|
|
некоторых случаях гем присоединен к ней |
||
|
ковалентной связью за счет радикалов |
||
|
цистеина, что характерно для цитохромов c1 |
||
|
и c. |
От двух атомов водорода, которые переносятся на комплекс III от KoQ, дальше по цепи транспортируются только электроны, два протона (H+)комплекс III выбрасывает в межмембранное пространство вместе с еще одной парой протонов, которые подхватываются комплексом из матрикса. Таким образом, комплекс III в сумме выбрасывает в межмембранное пространство 4 протона. Поэтому комплекс III, как и комплекс I, является протонным генератором, и целью его работы также является создание +.
КОМПЛЕКС IV.
Комплекс IV называется цитохромоксидазой. Он способен захватывать из матрикса 4 протона. Два из них он отправляет в межмембранное пространство, а остальные передает на образование воды.
Благодаря многоступенчатой передаче энергия в дыхательной цепи выделяется не мгновенно, а постепенно (маленькими порциями) при каждой реакции переноса. Эти порции энергии не одинаковы по величине. Их величина определяется разницей между ОВП двух соседних переносчиков. Если эта разница небольшая, то энергии выделяется мало — она рассеивается в виде тепла. Но на нескольких стадиях ее достаточно, чтобы синтезировать макроэргические связи в молекуле АТФ. Такими стадиями являются:
1)НАД/ФАД — разность потенциалов 0.25V.
2)Цитохромы b/cc1 — 0.18V
3)aa3/O-2 — 0.53V.
Значит, на каждую пару атомов водорода, отнятых от субстрата, возможен синтез 3-х молекул АТФ.
АДФ + Ф + ЭНЕРГИЯ ——-> АТФ + Н2О Макроэргическая связь — это такая ковалентная связь,
при гидролизе которой выделяется не менее 30 кДж/моль энергии. Эта связь обозначается знаком ~.
Синтез АТФ за счет энергии, которая выделяется в системе МтО, называется ОКИСЛИТЕЛЬНЫМ ФОСФОРИЛИРОВАНИЕМ. Основная роль АТФ —
обеспечение энергией процесса синтеза АТФ.
Для оценки эффективности работы системы МтО при окислении вычисляют КОЭФФИЦИЕНТ P/O. Он показывает, сколько молекул неорганического фосфата присоединилось к АДФ в расчете на один атом кислорода.
Для главной (полная) цепи Р/О=3 (10H+/2H+(затраты на освобождение АТФ из комплекса с ферментом) + 1H+ (затраты на транспорт фосфата)) = 3,3 (округляют до 3-х)), коэффициент полезного действия системы — 65%, для
7
укороченной P/O=2 (6H+/2H+(затраты на освобождение АТФ из комплекса с ферментом) + 1H+ (затраты на транспорт фосфата)) = 2, для максимально укороченной P/O=1 (4H+/2H+(затраты на освобождение АТФ из комплекса с ферментом) + 1H+ (затраты на транспорт фосфата)) = 1.
Система МтО потребляет 90% кислорода, поступающего в клетку. При этом в сутки образуется 62 килограмма АТФ. Но в клетках организма содержится всего 20-30 граммов АТФ. Поэтому молекула АТФ в сутки гидролизуется и снова синтезируется в среднем 2500 раз (средняя продолжительность жизни молекулы АТФ — полминуты).
ОСНОВНЫЕ ПРОЦЕССЫ, ДЛЯ КОТОРЫХ ИСПОЛЬЗУЕТСЯ ЭНЕРГИЯ АТФ:
1.Синтез различных веществ.
2.Активный транспорт (транспорт веществ через мембрану против градиента их концентраций). 30% от общего количества расходуемого АТФ приходится на Na+,К+-АТФазу.
3.Механическое движение (мышечная работа).
СИНТЕЗ АТФ.
Во внутренней мембране митохондрий расположен интегральный белковый комплекс – Н+—
зависимая АТФ-синтаза seu Н+-зависимая АТФ-аза (два разных названия связаны с полной обратимостью катализируемой реакции), обладающий значительной молекулярной массой – более, чем 500кДа. Состоит из двух субъединиц: FO и F1.
F1 представляет из себя грибовидный вырост на матриксной поверхности внутренней митохондриальной мембраны, FO же пронизывает эту мембрану насквозь. В толще FO расположен протонный канал, позволяющий протонам возвращаться обратно в матрикс по градиенту их концентраций.
F1 способна связывать АДФ и фосфат на своей поверхности с образованием АТФ — без затраты энергии, но обязательно в комплексе с ферментом. Энергия необходима лишь для освобождения АТФ из этого комплекса. Эта энергия выделяется в результате тока протонов через протонный канал FO.
В дыхательной цепи сопряжение абсолютно: ни одно вещество не может окисляться без восстановления другого вещества.
Но при синтезе АТФ сопряжение одностороннее: окисление может идти без фосфорилирования, а фосфорилирование без окисления никогда не идѐт. Это означает, что система МтО может работать без синтеза АТФ, но АТФ не может быть синтезирована, если не работает система МтО.
СПЕЦИФИЧЕСКИЕ ИНГИБИТОРЫ ТКАНЕВОГО ДЫХАНИЯ
К ним относятся вещества, прекращающие работу того или иного комплекса дыхательной
цепи.
Ингибитором комплекса I является яд растительного происхождения РОТЕНОН. Некоторые народности раньше использовали его в рыбной ловле.
Ингибиторами комплекса IV являются ЦИАНИДЫ, угарный газ СО, сероводород H2S.
ВЕЩЕСТВА-РАЗОБЩИТЕЛИ ПРОЦЕССОВ ОКИСЛЕНИЯ И ФОСФОРИЛИРОВАНИЯ
Они не прекращают процессов окисления, но снижают синтез АТФ. Дыхательная цепь работает, а АТФ при этом синтезируется в меньшем количестве, чем в норме. Тогда энергия, получаемая при переносе электронов по цепи МтО, выделяется в виде тепла. Такое состояние, когда происходит окисление субстратов, а фосфорилирование (образование АТФ из АДФ и Ф) не идет, называется РАЗОБЩЕНИЕМ ОКИСЛЕНИЯ И ФОСФОРИЛИРОВАНИЯ. К такому состоянию может приводить действие веществ-разобщителей:
2,4-ДИНИТРОФЕНОЛ, открытый в 1944 году Липманом, при введении в организм повышает температуру тела и понижает синтез АТФ. Это вещество, наряду с другими, открытыми позже, пытались использовать для лечения ожирения, но безуспешно.
Механизм действия веществ-разобщителей становится понятням только с точки зрения хемиоосмотической теории.
Разобщители являются слабыми кислотами, растворимыми в жирах. В межмембранном пространстве они связывают протоны, и затем диффундируют в матрикс, тем самым снижая .
Подобным действием обладает и йодсодержащие гормоны щитовидной железы – тироксин и трийодтиронин. При состояниях, сопровождающихся гиперфункцией щитовидной железы (например, Базедова болезнь), больным не хватает энергии АТФ: они много едят (нужно большое количество субстратов для окисления), но при этом теряют в весе. Большая часть энергии выделяется в виде тепла.
Схема цепи митохондриального окисления не раскрывает механизма образования АТФ путем окислительного фософорилирования. Этот механизм объясняется гипотезой П.Митчелла.

|
8 |
|||
|
ТЕОРИЯ СОПРЯЖЕНИЯ ОКИСЛЕНИЯ |
|||
|
И |
ФОСФОРИЛИРОВАНИЯ ПИТЕРА |
||
|
МИТЧЕЛЛА. |
|||
|
Известно, что через мембрану митохондрии |
|||
|
могут |
свободно |
проникать только |
небольшие |
|
незаряженные молекулы, а также |
|||
|
гидрофобные молекулы. Энергия, которая |
|||
|
выделяется при переносе электронов по |
|||
|
цепи |
МтО, приводит к переносу протонов (Н+) из |
||
|
матрикса |
митохондрии в межмембранное |
||
|
пространство. Поэтому на внутренней |
|||
|
мембране |
митохондрий |
образуется |
|
|
градиент |
концентраций протонов: в |
межмембранном пространстве Н+ становится много, а в матриксе остается мало. Образуется разность потенциалов 0.14V — наружная часть мембраны заряжена положительно, а внутренняя — отрицательно. Накопившиеся в межмембранном пространстве Н+ стремятся выйти обратно в матрикс по градиенту их концентраций, но митохондриальная мембрана для них непроницаема. Единственный обратный путь в матрикс для протонов — через протонный канал фермента АТФ-синтетазы, которая встроена (built-in) во внутреннюю мембрану митохондрий. При движении протонов по этому каналу в матрикс их энергия используется АТФ-синтазой для синтеза АТФ. Синтезируется АТФ в матриксе митохондрий.
После синтеза АТФ переносится в цитоплазму путем облегчѐнной диффузии по градиенту концентраций, поскольку основные процессы, в которых АТФ потребляется, протекают в цитоплазме.
Как происходит транспорт АТФ из митохондрий в цитоплазму?
Для этого используется специфический для АТФ транспортный белок — АТФ/АДФ-транслоказа. Это интегральный белок, локализован во внутренней мембране митохондрий.
Во внутренней мембране митохондрий есть белок-переносчик — АТФ/АТФ-транслоказа, который имеет 2 центра связывания: со стороны матрикса для АТФ, снаружи — для АДФ. При изменении конформации АТФ/АДФ-транслоказы АДФ переносится в матрикс, а АТФ — в межмембранное пространство, а затем — в цитоплазму, где используется.
Для образования АТФ в матрикс всѐ время должен поступать неорганический фосфат (Ф). Для этого во внутренней мембране митохондрий есть транспортная система, которая обеспечивает перенос фосфата в матрикс сопряженно с переносом Н+. Это белок-переносчик, который имеет 2 центра связывания: для Ф и Н+. Ф и Н+ вместе переносятся из межмембранного пространства в матрикс.
Известны некоторые вещества, которые способны разобщать процессы окисления и фосфорилирования, приводя тем самым к уменьшению коэффициента р/о. К ним относятся йодсодержащие гормоны щитовидной
железы (тироксин, трийодтиронин), а также некоторые ксенобиотики (например, 2,4-динитрофенол). Такие вещества известны под общим названием «РАЗОБЩАЮЩИЕ ЯДЫ». Как действуют веществаразобщители окисления и фосфорилирования? Они могут образовывать собственные протонные каналы во внутренней мембране митохондрий. Поэтому часть протонов, вместо того, чтобы идти обратно в матрикс по протонному каналу АТФ-синтетазы, уходит туда по каналам веществразобщителей. В результате АТФ образуется меньше, и часть энергии выделяется в виде тепла.
АВТОНОМНАЯ САМОРЕГУЛЯЦИЯ СИСТЕМЫ МИТОХОНДРИАЛЬНОГО ОКИСЛЕНИЯ
Если клетка организма находится в условиях покоя, то АТФ мало используется и накапливается. Поэтому снижается концентрация АДФ и Ф. В этих условиях АТФ-синтетаза уже не получает из цитоплазмы достаточно фосфата и АДФ для синтеза АТФ. Еѐ активность понижается, и скорость движения протонов из межмембранного пространства в матрикс по протонному каналу этого фермента тоже падает. Поэтому сохраняется высокий градиент концентраций протонов на внутренней мембране митохондрий. В этих условиях энергии переноса водорода по цепи митохондриального окисления уже не хватает для выталкивания Н+ из матрикса в межмембранное пространство. Перенос водорода по цепи МтО тормозится и прекращается окисление субстратов.
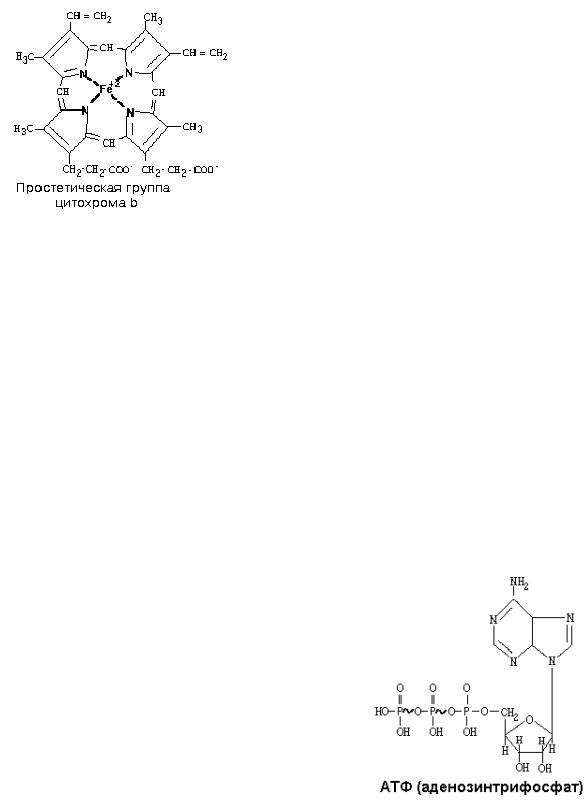
Метаболизм в клетке регулируется отношением АТФ/АДФ. Это отношение характеризует ЭНЕРГЕТИЧЕСКИЙ ЗАРЯД КЛЕТКИ.
9
В норме ЭЗК = 0.85-0.90. Может изменяться от 0 до 1. Высокий ЭЗК тормозит синтез АТФ, и активирует использование АТФ (АТФ——-> АДФ + Ф)
БИОЛОГИЧЕСКАЯ РОЛЬ МИТОХОНДРИАЛЬНОГО ОКИСЛЕНИЯ
Главная его функция — обеспечение организма запасами энергии в форме АТФ.
Именно митохондрии поставляют клетке большую часть необходимого ей АТФ.
В сутки синтезируется до 62 кг АТФ, хотя одновременно в организме никогда не бывает больше 30-40 граммов этого вещества. Т.е. наблюдается очень быстрое восстановление расходуемых молекул АТФ.
ВАРИАНТЫ ДЫХАТЕЛЬНОЙ ЦЕПИ.
1.ПОЛНАЯ ДЫХАТЕЛЬНАЯ ЦЕПЬ
Вэтой цепи окисляется небольшое количество субстратов, из которых главными являются четыре. Коэффициент Р/О=3.
|
СУБСТРАТЫ |
НИКОТИНАМИДНЫЕ ФЕРМЕНТЫ, ИХ |
|
ОКИСЛЯЮЩИЕ |
|
|
изолимонная кислота (изоцитрат) |
Изоцитратдегидрогеназа |
|
Яблочная кислота (малат) |
Малатдегидрогеназа |
|
Глутаминовая кислота (глутамат) |
Глутаматдегидрогеназа |
|
бета-гидроксиацил-КоА |
бета-гидроксиацил-КоА-дегидрогеназа |
Все ферменты полной цепи являются НАД-зависимыми дегидрогеназами.
2. СОКРАЩЕННАЯ (УКОРОЧЕННАЯ) ДЫХАТЕЛЬНАЯ ЦЕПЬ
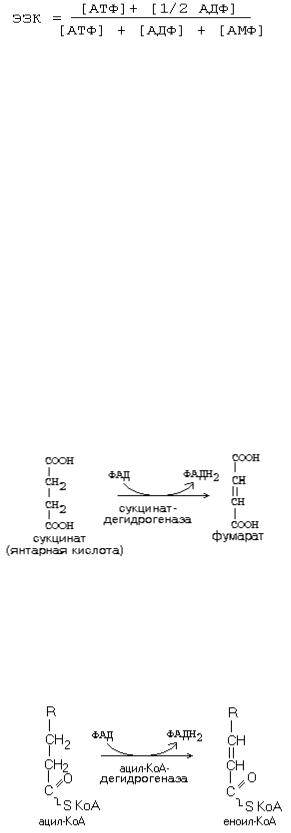
Известны два субстрата. Главным из них является сукцинат (янтарная кислота).
Сукцинатдегидрогеназа – это комплекс II, который в укороченном варианте цепи является начальным звеном окисления. В составе комплекса – простетическая группа ФАД и FeSII. От ФАД.Н2 два атома водорода переносятся на KoQ. Значит, первое звено, которое имеется в полной цепи — исключается. Перепад окислительно-восстановительного потенциала между ФАД и KoQ невелик. Поэтому переноса H+ в межмембранное пространство в этой точке не происходит. + также создается, но меньший, чем в полной цепи. Значит, меньше и эффективность фосфорилирования — коэффициент Р/О=2.
Аналогичным образом окисляется и второй субстрат – ацил-КоА (активная форма любой жирной кислоты):
3. МАКСИМАЛЬНО СОКРАЩЕННАЯ (МАКСИМАЛЬНО УКОРОЧЕННАЯ) ДЫХАТЕЛЬНАЯ ЦЕПЬ.
Она представлена только цитохромной частью. Эксперименты показали, что здесь может быть окислен только один субстрат — аскорбиновая кислота, с участием фермента, восстановленные эквиваленты включаются в цепь на уровне цитохрома С цитохромоксидазы (цитохром аа3), но в реальных условиях такого окисления практически не происходит. Образуется вода и 1 молекула АТФ. Коэффициент Р/О=1.
10
Возможность такого окисления доказана в эксперименте in vitro. А в живой клетке аскорбиновая кислота обычно используется как донор водорода в системе окисления оксигеназного типа (реакции, катализируемые монооксигеназами: смотрите лекцию «Внемитохондриальное окисление»). Такие реакции с участием витамина «С» особенно важны для формирования белка коллагена, в котором за счет монооксигеназной реакции образуется гидроксипролин.
ОКИСЛИТЕЛЬНОЕ ДЕКАРБОКСИЛИРОВАНИЕ ПИРОВИНОГРАДНОЙ И -КЕТОГЛУТАРОВОЙ КИСЛОТ В МИТОХОНДРИЯХ
Этот вариант дыхательной цепи удлинен по сравнению с полной цепью за счет того, что первое звено катализируется не никотинамидным ферментом, а мультиферментным комплексом. Это единая надмолекулярная структура. В состав этого комплекса входят 3 фермента и 5 коферментов. Такой комплекс называется мультиферментным комплексом окислительного декарбоксилирования -кетокислот, и он окисляет два субстрата:
1. Пировиноградную кислоту (пируват, ПВК)
Окисляется с помощью ферментов пируватдегидрогеназного комплекса; 2. -кетоглутаровую кислоту ( -КГ)
Окисляется с помощью ферментов -кетоглутаратдегидрогеназного комплекса.
Оба комплекса ферментов работают одинаково. Они катализируют реакции окислительного декарбоксилирования соответствующей -кетокислоты.
ОКИСЛИТЕЛЬНОЕ ДЕКАРБОКСИЛИРОВАНИЕ ПИРУВАТА
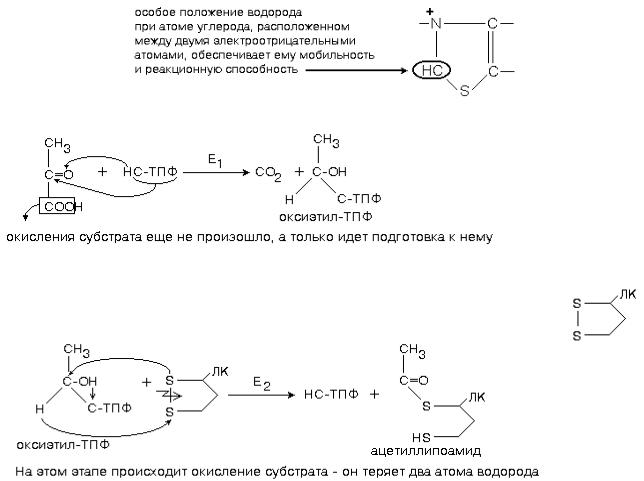
1-ю реакцию катализирует фермент ПИРУВАТДЕКАРБОКСИЛАЗА (Е1).
Простетической группой пируватдекарбоксилазы является тиаминдифосфат (ТПФ, тиаминпирофосфат, ТДФ) — это активная форма витамина В1. Активная часть ТПФ — тиазоловое кольцо и атом водорода в нем. Для краткости записывают: НС-ТПФ.
Пируватдекарбоксилаза отщепляет CO2, а оставшаяся оксиэтильная группа присоединяется к
ТПФ.
2-ю и 3-ю реакцию процесса катализирует фермент АЦИЛТРАНСФЕРАЗА (Е2). Простетическая группа ацетилтрансферазы — амид липоевой кислоты. Катализирует перенос оксиэтильного остатка на свой собственный кофермент (строение ЛК знать описательно по учебнику). В ее составе есть дисульфидная связь.
На этом этапе уже произошло окисление оксиэтильного остатка до остатка уксусной кислоты, одновременно с этим началось восстановление липоевой кислоты.
3 этап — продолжает работать фермент ацилтрансфераза.
- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Исрапилова А.И.
1
Османова П.М.
1
Гаджиева А.К.
1
Магомедова К.М.
1
1 Дагестанский государственный университет
За последние десятилетия было накоплено большое количество новых сведений, которые позволяют с новой стороны взглянуть на общепринятые модели, касающиеся митохондрий, их функций и строения. И в данном сообщении были собраны все основные новые открытия, которые позволят посмотреть на митохондрии с новой стороны.
митохондрии
атф
кальциевый обмен
апоптоз
фософорилирование
1. Бонь.Е.И. Медико-биологические и фундаментальные исследование/ Е.И.Бонь, Н.Е.Максимович//Оренбургский медицинский вестник, том 2 №1(25) -2015
2. Бра М. Митохондрии в программированной гибели клетки: различные механизмы гибели/М. Бра, Б. Квинан, С.А. Сузин // Биохимия. – 2005. – Т. 70, № 2. – С. 284–293.
3. Владимиров Ю.А. Дизрегуляция проницае мости мембран митохондрий, некроз и апоптоз / Ю.А. Владимиров // Дизрегуляционная патология. – М.: Медицина, 2002. – C. 127–156.
4. Aradjomande S.L.A. Newcomers in the process of mitochondrial permeabilisation / S.L. A. Aradjomande, J.C. Martinou // J. Cell Scien. – 2005. – Vol. 118. – P. 473–483
5. Becker T., Gebert M., Pfanner N., Laan M. Biogenesis of mitochondrial membrane proteins // Current Opinion in Cell Biology. – 2009. – № 21. – P. 484-493
6. Bhosale G., Sharpe J., Sundier S., Duchen M. Calcium signaling as a mediator of cell energy demand and a trigger to cell death // Annals of the NewYork academy of sciences. – 2015. – Vol. 1350. – P. 107-116
7. Bonifaz L., Cervantes-Silva M., Oniveros-Dotor E., Lopez-Villegas E., Sanchez-Garcia F. A role for mitochondria in antigen processing and presentation // Immunology. – 2015. – Vol. 144, № I.3. – P. 461-471.
8. Booth D., Joseph S., Hajnoczky G. Subcellular ROS imaging methods relevance for study of calcium signaling // Cell calcium. – 2016. – Vol. 60. – P. 65-73.
9. Buja L.M. Myocardial ischemia and reperfusion injury / L.M. Buja // Cardiovasc. Pathol. – 2005. – Vol. 14. – P. 170–175.
10. Cloonan S.M., Choi A. Mitochondria: sensors and mediators of innate immune receptor signaling // Current Opinion in Microbiology. – 2013. – № 16. – P. 1-12.
11. Crow M.T. Hypoxia, BNip3 proteins, and the mitochondrial death pathway in cardiomyocytes / M.T. Crow // Circ. Res. – 2002. – Vol. 92. – P. 183–185.
12. Csords G., Vrnai P., Golenar T., Sheu-Shing S., Hajnуczky G. Calcium transport across the inner mitochondrial membrane: Molecular mechanisms and pharmacology // Molecular and Cellular Endocrinology. – 2012. –№ 353. – P. 109-113.
13. Dupont G. Modeling the intracellular organization of calcium signaling // Wiley Periodicals. – 2014. – Vol. 6. – P. 227-237.
14. Erazo-Oliveras, A. Proteindelivery into live cells by incubationwithanendosomolyticagent /А. Erazo-Oliveras // Nat Methods. – 2014. – V. 56. – Р. 112-118.
15. Giorgi C., Agnoletto A., Bononi A., Bonora M., Marchi E.D., Marchi S., Missiroli S., Patergnani S., Poletti F., Rimessi A., Suski J.M., Wieckowski M.R., Pinton P. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine // Mitochondrion. – 2012. – № 12. – P. 77-85.
16. Guimaraes C.A. Programmed cell death: apoptosis and alternative deathstyles / C.A. Guimaraes, R. Linden // Eur. J. Biochem. – 2004. – Vol. 217. – P. 1638–1650.
17. Joseph N., Reicher B., Barda-Saad M. The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux // Biochemica et Biophysica. – 2014. – A. 1838. – P. 557-568
18. Kaufman R. J., Malhotra J. D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics // Biochimica et Biophysica Acta. – 2014. – Vol. 1843. – P. 2233-2239
19. Kock R., Josefsan C., Hill G. Mitochondrial functions, ornamentation and immunocompetence // Biol. Rev. – 2016. – Vol. 456. – P. 1-12.
20. Levine B. Autophagy in cell death: an innocent convict? / B. Levine, J. Yuan // J. Clin. Invest. – 2005. – Vol. 115. – P. 2679– 2688.
21. Logan D.C. The mitochondrial compartment // J. Exp. Bot. 2006. V. 57. P. 1225-1243
22. Role of the mitochondrial permeability transition in myocardial disease / J.N. Weiss, P. Korge, H.M. Honda et al. // Circ. Res. – 2003. – Vol. 93. – P. 292–301
23. Serricchio, M. Cardiolipin synthesizing enzymes form a complex that interacts with cardiolipin-dependent membrane organizing proteins / Serricchio M., Vissa A., Kim P. K., YipC. M., McQuibbanG. A. // Acta Molecular Cell Biology Lipids. – 2018. – V. 4. – P. 447-457
24. Xu C. Endoplasmic reticulum stress: cell life and death decisions / C. Xu, B. BaillyMaitre, J.C. Reed // J. Clin. Invest. – 2005. – Vol. 115. – P. 2656–2664.
Митохондрии представляют собой внутриклеточные органеллы эукариот, основной функцией которых является выработка АТФ в результате реакции окислительного фосфорилирования. (Logan, 2006)
Каждая митохондрия содержит высокоспециализированные мембраны, играющие ключевую роль в ее активности. Мембраны образуют два изолированных митохондриальных компартмента: внутренний матрикс и узкое межмембранное пространство. Каждый отдел содержит уникальный набор белков. В состав наружной мембраны входит белок порин, который образует широкие гидрофильные каналы в липидном бислое. (Максимович, 2015). В результате эта мембрана напоминает сито, проницаемое для всех молекул массой менее 10000 дальтон, в том числе низкомолекулярных. Эти молекулы могут проникать в межмембранное пространство, но большая их часть не способна проходить через непроницаемую внутреннюю мембрану. Основная функциональная часть митохондрии– матрикс и окружающая его внутренняя мембрана. Внутренняя мембрана содержит большое количество «двойного» фосфолипида кардиолипина (30%), что обеспечивает непроницаемость мембраны для ионов и отличается необычно высоким содержанием белка (около 70% от веса). Многие из белков являются компонентами электронтранспортной цепи, поддерживающей протонный градиент на мембране. Другой большой белковый комплекс–фермент АТФ-синтаза, катализирующий синтез АТФ, через который протоны возвращаются в матрикс по электрохимическому градиенту (Erazo-Oliveras,2014).
Во внутреннюю митохондриальную мембрану встроены ферменты дыхательной цепи, необходимые для процесса окислительного фосфорилирования, образующего основную часть АТФ, и транспортные белки, обусловливающие ее избирательную проницаемость. Внутренняя мембрана митохондрий непроницаема для Н+, ОН—, всех анионов и катионов. Транспорт необходимых веществ и неорганических ионов происходит при участии белков-переносчиков (Serricchio, 2018). Матрикс митохондрий имеет более вязкую консистенцию по сравнению с цитоплазмой клетки. В нем находятся ферменты, митохондриальная ДНК, рибосомы, органические соединения, ионы, соли кальция и магния. Ферменты, расположенные в матриксе, участвуют в цикле Кребса, окислительном фосфорилировании, окислении пирувата и бета-окислении жирных кислот. Субстратом для окислительного метаболизма в митохондриях служат главным образом жирные кислоты и пируват, образуемый в результате гликолиза в цитозоле. Эти вещества избирательно транспортируются из цитозоля в митохондриальный матрикс, где распадаются до двухуглеродных групп, присоединенных к ацетилкоферменту А (ацетил-СоА). В составе молекулы ацетил-СоА каждая ацетильная группа поступает в цикл Кребса для дальнейшего расщепления, где при окислении двухуглеродных атомов ацетил-СоА происходит извлечение высокоэнергетических электронов.
Роль митохондрий в энергетике клетки
Наиболее характерной особенностью митохондрий является содержание в них большого числа ферментов, участвующих в аэробном «дыхании». Большая часть энергии, которая освобождается при переносе электронов, аккумулируется в макроэргических фосфатных связях АТФ. (Максимович, 2015)
Окисление ацетильной группы в цикле Кребса ведет к образованию молекул восстановленного NADH и восстановленного FADH2. Вначале почти вся энергия, получаемая на ранних этапах окисления питательных веществ, аккумулируется в форме высокоэнергетических электронов NADH и FADH2. NADH, компонент NADH-дегидрогеназного комплекса, образовавшийся в цитозоле при гликолизе, передает свои электроны в дыхательную цепь. Так как NADH не способен проходить через внутреннюю мембрану, перенос электронов от него осуществляется непрямым путем при помощи одной из челночных систем, транспортирующих в митохондрию карнитин, который после окисления возвращается в цитозоль с последующим его восстановлением с помощью NADH. Другой субстрат, FADH2 передает свои электроны в дыхательную цепь непосредственно. Электроны этих субстратов восстанавливают молекулярный кислород (акцептор электронов) в дыхательной цепи с образованием метаболической воды. Так как большое количество высвобождаемой энергии используется ферментами внутренней мембраны для образования АТФ из AДФ, эти реакции называют окислительным фосфорилированием. На внутренней мембране создается электрохимический протонный градиент. Митохондриальная дыхательная цепь внутренней мембраны способна перемещать протоны Н+. При прохождении электронов по дыхательной цепи происходит их «откачивание» из матрикса. АТФ-синтаза может использовать энергию гидролиза АТФ для переноса Н+ через мембрану, а при достаточно большом протонном градиенте протоны начинают «течь» через фермент в обратном направлении, что сопровождается синтезом АТФ. Все белки-переносчики электронов группируются в 4 больших комплекса дыхательных ферментов, каждый из которых содержит трансмембранные белки, прочно закрепляющие комплекс во внутренней мембране митохондрии. Комплекс I (NADH-убихиноноксидоредуктаза; NADH-дегидрогеназа), комплекс II (сукцинатдегидрогеназа; сукцинат-убихинон оксидоредуктаза), комплекс III (комплекс цитохромов b, c1; убихинон-цитохром c оксидоредуктаза), комплекс IV (цитохром c оксидаза; цитохромоксидаза; цитохром с-O2 оксидоредуктаза). Каждый последующий комплекс обладает большим сродством к электронам, чем предыдущий. (Logan, 2006) Электроны последовательно переходят от одного комплекса на другой, пока не восстановят кислород, являющийся их акцептором.(Максимович, 2015)
Синтез АТФ – не единственный процесс, идущий за счет энергии электрохимического градиента. В матриксе, где находятся ферменты, участвующие в цикле Кребса и других метаболических реакциях, необходимо поддерживать высокие концентрации различных субстратов. Поэтому через внутреннюю мембрану должны транспортироваться различные несущие заряд субстраты. Их активно перекачивают против электрохимических градиентов встроенные в мембрану белки-переносчики. Энергия электрохимического протонного градиента используется также для переноса в матрикс ионов Са2+, которые играют важную роль в регуляции активности некоторых митохондриальных ферментов. Большое значение имеет поглощение митохондриями этих ионов для удаления их из цитозоля, где высокая концентрация Са2+ является опасной. (Cloonan 2013)
Роль митохондрий в кальциевом гомеостазе
Центральным механизмом в реализации иммунного ответа является кальциевая сигнализация. Иммунореактивность лимфоцитов обеспечивается интеграцией митохондрий и механизмов кальциевой сигнализации. Митохондрии играют важную роль в гомеостазе Ca2+ лимфоцитов, как и в других клетках. Они имеют огромный потенциал для его быстрого накопления, поэтому участвуют в модуляции пространственно-временного профиля кальциевых сигналов (Bonifaz 2015, Chandel 2014).
В последние годы все большее внимание исследователей привлекает изучение работы митохондрий как кальциевых депо клетки в процессе реализации специфических функций иммунокомпетентных клеток, так как белки компоненты этой сложной системы регуляции кальциевого гомеостаза могут рассматриваться в качестве молекул-мишеней для направленной регуляции функциональной активности лимфоцитов в норме и при патологических процессах (воспаление, аутоиммунная патология, аллергические реакции, иммунодефициты).
Взаимосвязь между динамикой митохондрий и кальциевыми сигналами связана с двумя аспектами клеточных функций: хемотаксисом и регуляцией формирования иммунологического синапса. Иммунологический синапс представляет собой важный кальций – зависимый процесс, который осуществляется во время активации лимфоцитов (Kock 2016). Особая роль при этом принадлежит митохондриям, так как способность митохондрий поглощать Ca2+ оказывает влияние на формирование кальциевых сигналов и их распространение. Во время активации лимфоцитов митохондрии локализуются вблизи иммунологического синапса, образуют сложный структурный комплекс, связывающий мембраны эндоплазматического ретикулума (ЭПР) с мембранами митохондрий и плазматической мембраной, так называемую «ассоциированную мембрану» (Quintanaa 2012). Поглощая ионы из внутриклеточной среды, данные органеллы, накапливают их в матриксе митохондрий и высвобождают избыточное количество в цитозоль. Так митохондрии регулируют уровень Ca2+, действуя в качестве буферов, активируют или ограничивают действие кальциевых сигналов в клетке, а именно, контролируют уровень Ca2+ в цитозоле и цитоплазматических микродоменах, изменяя частоту осцилляций кальциевых сигналов и снижая амплитуду распространяющихся волн (Dupont 2014).
Стабильный уровень Ca2+ в митохондриях сохраняется в результате равномерного накопления ионов и их высвобождении при значительном повышении уровня Ca2+ в матриксе, за счет слаженной работы транспортной системы внешней и внутренней мембран митохондрий. Данная система включает основной канал тока Ca2+ через наружную мембрану – потенциал-зависимый анионный канал; также систему унипорта внутренней мембраны и его молекулярные компоненты, регулирующие активность; два пути высвобождения Ca2+ в цитозоль – H+/Ca2+ насос и проницаемая пора мембраны митохондрий. Ток Ca2+ через потенциал – зависимый канал и систему унипорта осуществляется за счет электрохимического протонного градиента (Kaufman 2014).
Изучение способности митохондрий поглощать Ca2+ показало, что аккумуляция Ca2+ осуществляется за счет градиента разности потенциалов (∆ψ), энергии, образующегося при окислении субстратов или активности H+АТФазы. Отношение уровня Ca2+ при равновесии (∆µCa = 0) составляет [Ca2+]мит / [Ca2+]цит = 106 , с учетом мембранного потенциала и заряда иона.
Так, в цитозоле уровень кальция составляет в норме 10-7М, тогда как в матриксе – 10-1М. Однако для митохондрий такое значение не совместимо с их жизнедеятельностью, поэтому Ca2+ высвобождается через систему антипорта при значительном повышении в матриксе. Митохондрии поглощают Ca2+ если уровень ионов в цитозоле превышает 400 нм. Ток Ca2+ из матрикса в электроневозбудимых клетках зависит от механизма H+/Ca2+ обмена. Система антипорта H+/Ca2+ 7 представляет собой высококонсервативный трансмембранный белок, экспрессируемый внутренней мембраной митохондрии, который контролирует уровень кальция в митохондриях.
Были определены белки, участвующие в контроле Ca2+ тока сквозь внутреннюю мембрану митохондрий (Becker 2009). В частности, в 2010 г. были исследованы Na+/Ca2+ насосы; белки – регуляторы поглощения Ca2+ митохондриями, они получили название mitochondrial calcium uptake 1 белки –MICU1; затем были обнаружены и частично охарактеризованы потенциальные регуляторы тока Ca2+ в митохондрии: MICUb, MICU2, MICU3, EMRE. На основании проведенных исследований сложилась более четкая картина осуществления поглощения ионов кальция митохондриями и сохранении гомеостаза Ca2+ как внутри органеллы, так и клеточной системе, в целом (Becker 2009).
Внутренняя мембрана митохондрий является непроницаемой для кальция и требует специфических транспортеров для переноса Ca2+ из цитозоля в митохондрии.(Giorgi 2012). Транспорт Ca2+ в матрикс митохондрий через внутреннюю мембрану осуществляется двумя механизмами: 1) локализацией митохондрий вблизи ЭПР с образованием высококонцентрированного микродомена; 2) захватом кальция низкоаффинным транспортером-унипортом. Эти два механизма дополняют друг друга (Csords 2012). Повышение уровня кальция в митохондриях достигает высоких значений, на два порядка выше, чем в цитозоле. Это обеспечивается кальций-селективным каналом или унипортом внутренней мембраны митохондрий. Кальциевый унипорт митохондрий (mitochondrial calcium uniporter-MSU) является белком, экспрессирующимся на внутренней мембране митохондрий с большим электрохимическим градиентом. Это довольно высокоселективный канал, но с низкой аффинностью к ионам Ca2+. Свойства унипорта зависят от активности его регуляторных субъединиц: MICU1и MICU2 (Giorgi 2012).
Существует зависимость между уменьшением расстояния между митохондриями и плазматической мембраной и увеличением открытия кальциевых каналов в мембране ЭПР. Тесный контакт между плазматической мембраной, ЭПР и митохондриями в иммунологическом синапсе дает возможность митохондриям быстро захватывать большое количество Ca2+ из внеклеточной среды (Kaufman 2014). Во время образования ассоциированной мембраны между митохондриями и ЭПР осуществляется поглощение Ca2+ митохондриями, в состоянии покоя поглощение Ca2+ подавляется, вследствие предотвращения перегрузки Ca2+ в матриксе, рассеяния разности потенциалов и снижения синтеза АТФ, и, напротив, в стимулированном состоянии митохондрии активно поглощают Ca2+, в этом случае наблюдается экспоненциальные повышение уровня ионов (Bhosale 2015).
Шапероны в мембранах ЭПР и митохондрий обеспечивают физическое и функциональное взаимодействие между ЭПР и митохондриями. В формировании АММ главную роль играет глюкозо-регулирующий белок – шаперон GRP75, который содержится в большом количестве в митохондриях. Этот шаперон контролирует передачу кальциевого сигнала от ЭПР к митохондриям и индуцирует взаимодействие между фосфоинозитол3-фосфат-чувствительными рецепторами и VDAC1. В этом случае шаперон образует между мембранами ЭПР и митохондрий туннель для Ca2+, позволяя более эффективно проникать ионам из ЭПР во внешнюю мембрану митохондрий.
Митохондриальная пора – канал, для которого характерны два состояния: низкокондуктивное и высококондуктивное. В первом случае канал открывается временно, во втором случае пора полностью и необратимо открыта, вследствие этого нарушается ток H+. В результате происходит разобщение окислительного фосфорилирования, которое предотвращает синтез АТФ. Открытие этого канала приводит к гибели клетки вследствие развития следующих событий: деполяризации мембраны, в результате разобщения окислительного фосфорилирования и продукции АТФ; набухание митохондрии, вследствие изменения осмолярности; высвобождение в цитозоль проапоптогенных белков (Booth 2016).
Роль митохондрий в апоптозе
Установлено, что основным компонентом, осуществляющим восприятие стимулов ПГК и активизирующим механизмы реализации той или иной формы ПГК, являются митохондрии. Предполагается, что на уровне митохондрий осуществляется интеграция сигналов активизирующих и подавляющих процесс ПГК, следствием чего является дальнейшая реализация программированной клеточной гибели или ее подавление.
На сегодняшний день показано существование трех основных форм программированной гибели клетки: апоптоз (I тип ПГК), аутофагия (II тип ПГК), некрозоподобная ПГК (III тип ПГК). Каждый из этих типов гибели клетки характеризуется собственными биохимическими, молекулярными и морфологическими особенностями (Бра 2005).
При апоптозе наблюдается уменьшение клетки в объеме, конденсация хроматина и фрагментация ДНК на олигонуклеосомные фрагменты. Митохондрии и рибосомы во время реализации апоптоза сохраняют в основном свою структуру и частично – функции. Заключительный этап апоптоза характеризуется разрушением цитоскелета, что приводит к сморщиванию клетки и ее фрагментации на апоптотические тельца, поглощаемые макрофагами или другими соседними клетками.
Ключевыми участниками терминальной фазы апоптотической программы являются цистеиновые протеазы – каспазы, осуществляющие деградацию белковых структур клетки и активирующие нуклеазы. (Бра 2005). Для аутофагии характерно набухание митохондрий и цистерн эндоплазматического ретикулума, увеличение аппарата Гольджи, секвестрация клеточных органелл аутофагическими вакуолями, конденсация хроматина и коллапс ядра.
Терминальным этапом аутофагии является разрушение клеточных органелл лизосомальными ферментами, следствием чего является деградация клетки. Образующийся после реализации аутофагии клеточный дебрис поглощается соседними клетками (Levine 2005). Заключительным событием в этом процессе является разрыв плазматической мембраны, способствующий излиянию содержимого клетки в межклеточное пространство, что способствует индукции воспалительной реакции.
Соотношение различных типов ПГК может варьироваться в зависимости от типа и силы воздействия стимула, активизирующего ПГК.
Важной особенностью митохондрий является способность к значительной амплификации исходящих от них стимулов, активирующих ПГК. Показано, что открытие митохондриальных пор является общим моментом в реализации механизмов всех обсуждаемых выше форм ПГК (Владимиров 2002). Образование пор в митохондриях приводит к выходу из митохондрий цитохрома С, способствующего образованию апоптосомы и активирующего каспазы. Этот процесс является основным механизмом апоптотической гибели клетки. Через открытые поры в митохондриях в цитоплазму высвобождаются также факторы, перемещающиеся в ядро и активирующие реализацию ПГК по независимым от каспаз механизмам: эндонуклеаза G и AIF, связывающий ДНК и активирующий нуклеазы и протеазы в ядре. Показано, что данные факторы принимают участие в развитии как апоптоза, так и некроза. Помимо активаторов ПГК, митохондрии также высвобождают ингибиторы белков, блокирующих ПГК (Smac/DIABLO, Omi/ HtrA2) и предшественников каспаз (прокаспаза 2, 3, 9) (Бра 2005).
К небелковым медиаторам клеточной гибели относятся ионы Ca2+, активирующие при их выходе в цитоплазму кальпаины и Ca2+зависимые липазы, что приводит к реализации некротической формы ПГК. Дополнительным фактором индукции ПГК является увеличение продукции компонентами дыхательной цепи митохондрий активных форм кислорода, активирующих механизмы апоптоза, аутофагии и некроза. На сегодняшний день известны митохондриальные апоптотические поры (mitochondrial apoptotic pores – MAP) и поры повышенной проницаемости или мегаканалы (permeability transition pores – РТP). Механизмом образования апоптотических пор в митохондриях является олигомеризация на митохондриальной мембране белков Bax и Bak. (Aradjomande 2005).
Процесс формирования пор в митохондриях находится под жестким контролем различных регуляторных систем клетки. Установлено, что образованию MAP за счет олигомеризации Bax и Bak способствуют белки индукторы ПГК семейства Bcl 2: Bax, Bak, Bok, Boo, Bcl G, Bcl B, Bcl rambo, Bad, Bim, Bmf, Bid, Noxa, Puma, BNip3. Установлено, что BNip3 и активная поли(АДФ-рибоза)полимераза-1 (PARP-1) индуцируют открытие PTP, что сопровождается снижением митохондриального потенциала и увеличением продукции активных форм кислорода (Crow 2002) .Образование митохондриальных пор возможно посредством воздействия на митохондрии цитоплазматических ионов Ca2+ и активных форм кислорода, что приводит к ПОЛ или липолизу, опосредованного фосфолипазой А2. Установлено, что повреждение митохондрий и развитие их функциональных нарушений при различных патологических процессах и токсических повреждениях данных органелл также способствует активизации ПГК посредством обсуждаемых выше механизмов(Владимиров 2002).
Существует мнение, что «выбор» клеткой активизации механизмов той или иной формы программированной гибели определяется количеством открытых пор в митохондриях. В том случае, если PTP формируются в нескольких митохондриях, в клетке активируется процесс аутофагии. Когда PTP открываются у большего числа митохондрий, в клетке инициируется апоптоз, что, вероятно, является следствием увеличения в цитоплазме количества цитохрома С и AIF. Наконец, когда в клетке практически во всех митохондриях открываются РТP, происходит разобщение окисления и фосфорилирования и интенсивный гидролиз АТФ митохондриальной АТФ-азой, активизируются механизмы некрозоподобной клеточной гибели (Guimaraes 2004). Минимальное количество открытых пор принципиально не влияет на процесс клеточной гибели, при большем количестве.
Незначительное увеличение концентрации ионов кальция в цитоплазме приводит к развитию апоптоза, в то время как существенное возрастание их уровня индуцирует некроз. К возможным механизмам этого процесса относят воздействие ионов кальция на митохондрии и активацию Ca2+-зависимых протеаз (кальпаинов) (Crow 2002). Установлено, что увеличение внутриклеточной концентрации Ca2+ сопровождается открытием РТP в митохондриях и снижением митохондриального трансмембранного потенциала, следствием чего является активация программированной гибели клетки (Владимиров 2002). Известно, что увеличение внутриклеточной концентрации ионов Ca2+ способствует активации кальпаинов, Ca2+-зависимых фосфолипаз и нуклеаз, приводящих к разрушению внутриклеточных структур и реализации ПГК по механизму некроза (Endoplasmic 2005).
Считается, что определенное значение в реализации апоптоза и некрозоподобной ПГК имеет уровень продукции АТФ. Известно, что при низком уровне АТФ в клетке протекает процесс программированной гибели клетки по механизму некроза, достаточное энергообеспечение клетки способствует прохождению ПГК по механизму апоптоза (Buja 2005).
Установлено, что митохондрии обладают широким спектром белковых (цитохром С, эндонуклеаза G, AIF,) и небелковых факторов (ионы Ca2+, активные формы кислорода), активизирующих процесс клеточной гибели после высвобождения их в цитоплазму. В настоящее время существует аргументированная гипотеза, предполагающая, что накопление нарушений в митохондриальном геноме и прогрессирование митохондриальной дисфункции является одним из механизмов старения организма и развития различных патологических процессов.
На сегодняшний день известны митохондриальные апоптотические поры (MAP) и поры повышенной проницаемости или мегаканалы (permeability transition pores – РТP). Механизмом образования апоптотических пор в митохондриях является олигомеризация на митохондриальной мембране белков Bax и Bak. PTP формируются за счет объединения в единый комплекс АТФ –АДФ- антипортера, локализованного во внутренней митохондриальной мембране, циклофилина D, находящегося в матриксе митохондрий, и порина (voltage dependent anion channel, VDAC) – ионного канала внешней митохондриальной мембраны (Aradjomande, 2005).
В митохондриях в результате «утечки» электронов их электронтранс-портной цепи генерируется супероксиданион. При нарушение баланса между системами генерации и системами удаления АФК в митохондриях наблюдается возникновение окислительного стресса, что приводит к открытию неспецифической поры. Это обуславливает потерю мембранного потенциала, и, следовательно, невозможность импортирования митохондриальных белков, которые синтезируются в цитозоле. Свoбoдные рaдикaлы и рeaкции с их участием в последние годы стали причиной старения и возникновения многих заболеваний человека. Важным процессом в запуске митохондриального пути гибели клеток являются нарушения трансмембранного потенциала митохондрий вследствие повышения концентрации Са2+ в саркоплазматическом ретикуломе. Повышение уровня Са2+ внутри митохондрий способствует появлению транзиторных пор во внутреннем слое мембран митохондрий, что является причиной изменения электролитного баланса и нарушения энергетического потенциала его мембраны. Кроме того, избыточные ионы кальция нарушают функцию митохондрий, обеспечивающую поток электронов от НАДФ к кислороду и одновременно перенос протонов от матрикса митохондрий в межмембранное пространство, что определяет формирование электрохимического потенциала на внутренней мембране митохондрий. Дефицит в образовании НАДФ, с одной стороны, приводит к прекращению синтеза аденозинтрифосфата (АТФ), с другой — энергия, полученная от распада АТФ, используется для восстановления нарушенного электрического потенциала мембраны митохондрий. В результате митохондрии перестают быть источником энергии клеток и начинают ее поглощать. Истощение энергетических ресурсов клетки является причиной продолжения накопления Са2+ и воды в митохондриях, что приводит к появлению проницаемых пор (mPTP) во внешнем слое митохондриальных мембран (Kaufman 2014). Считается, что в открывании проницаемых пор играет важную роль также митохондриальный оксид азота (NO). Предполагается, что активный метаболит NO — пероксинитрит — может проникать в митохондрии из сарколеммы, а также вырабатываться в митохондриях под воздействием повышения активности Ca2+-чувствительных митохондриальных синтаз NO NOS (mNOS) (Kaufman 2014). Однако, что является источником образования NO в митохондриях, остается неясным, и требуются дальнейшие исследования в этом направлении. Высокая концентрация NO в митохондриях необратимо повреждает ряд компонентов дыхательной цепи и ингибирует продукцию АТФ. Помимо этого NO, путем образования транзиторных пор во внешней митохондральной мембране индуцирует апоптоз. В отличие от индуцибельной NO-синтазы (iNOS) митохондриальная NO-синтаза является кальцийзависимой, т.е. ее активность регулируется уровнем Ca2+ (Aradjomande 2005).
Таким образом, Митохондриальный путь апоптоза предусматривает не только активацию каспаз, но и доставку в ядро клетки активных ферментов — эндонуклеазы G и апоптозиндуцирующего фактора, способных вызвать деградацию генетического материала без активации каспаз (Kaufman 2014).
Библиографическая ссылка
Исрапилова А.И., Османова П.М., Гаджиева А.К., Магомедова К.М. СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯ О РОЛИ МИТОХОНДРИЙ В ФУНКЦИОНИРОВАНИИ КЛЕТКИ // Международный студенческий научный вестник. – 2020. – № 5.
;
URL: https://eduherald.ru/ru/article/view?id=20282 (дата обращения: 22.03.2023).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)
Митохондриальное окисление. Функции митохондрий
Митохондриальное окисление — мультиферментная система, постепенно транспортирующая протоны и электроны на кислород с образованием молекулы воды.
Все ферменты митохондриального окисления встроены во внутреннюю мембрану митохондрий. Только первый переносчик протонов и электронов — никотинамидная дегидрогеназа расположена в матриксе митохондрии. Этот фермент отнимает водород от субстрата и передает его следующему переносчику. Полный комплекс таких ферментов образует «дыхательный ансамбль» («дыхательную цепь»), в пределах которого атомы водорода отнимаются от субстрата, затем передаются последовательно от одного переносчика к другому, и, наконец, передаются на кислород воздуха с образованием воды.
Существует строгая последовательность работы каждого звена в цепочке переносчиков. Эта последовательность определяется величиной редокс-потенциала (окислительно-восстановительный — ОВП) каждого звена. ОВП — это химическая характеристика способности вещества принимать и удерживать электроны. Выражается в вольтах (V). Вещества с положительным ОВП окисляют водород (отнимают от него электроны), вещества с отрицательным ОВП окисляются самим водородом. Самый низкий ОВП имеет начальное звено цепи, самый высокий — у кислорода, расположенного в конце цепочки переносчиков. Таким образом, передача водорода идет от более низкого к более высокому ОВП. Перенос водорода и электронов возможен только в одном направлении — в порядке возрастания их ОВП: от -0.32V у никотинамидных дегидрогеназ (первого компонента главной цепи МтО) до 0.82V у О2, обладающего самым высоким редокс-потенциалом.
На одной из стадий происходит разделение атомов водорода на Н+ и электроны. Протоны остаются временно в окружающей среде, а электроны идут дальше по цепи и в ее конце используются для активации О2. Кислород является конечным акцептором электронов.
O2 + 4e → 2O2 (полное восстановление кислорода)
Все реакции, происходящие в дыхательной цепи, сопряжены. Переносчики водорода и электронов расположены в строгом порядке, в соответствии с величиной их редокс-потенциала.
В настоящее время различают три варианта дыхательных цепей:
- Главная (полная) цепь
- Укороченная (сокращенная) цепь
- Максимально укороченная цепь
Митохондриальное окисление. ГЛАВНАЯ ДЫХАТЕЛЬНАЯ ЦЕПЬ
Главная дыхательная цепь — это три мультиферментных комплекса, встроенных во внутреннюю мембрану митохондрии. Обозначаются они латинскими цифрами – I, III и IV.
СХЕМА ГЛАВНОЙ (ПОЛНОЙ) ДЫХАТЕЛЬНОЙ ЦЕПИ МИТОХОНДРИАЛЬНОГО ОКИСЛЕНИЯ

DmH+ — положительная величина. Его можно выразить как в вольтах (V), так и в единицах энергии (кДж/моль). Изменение значения pH на одну единицу соответствует 0,06V или 5,7 кДж/моль.
Энергия DmH+ используется для следующих процессов:
- Синтез АТФ.
- Получение тепла (особенно важно для бурого жира и для мышечной ткани птиц).
- Выполнение осмотической работы (транспорт фосфата в матрикс митохондрии).
- Мышечная работа (в некоторых случаях).
- Для человека наиболее важен синтез АТФ.
В полной цепи при окислении субстрата два атома водорода переносятся на НАД – кофермент никотинамидных дегидрогеназ.
Как видно из приведенной схемы, в полной цепи при передаче двух атомов водорода на кислород воздуха, в межмембранном пространстве оказываются 10 протонов, перенесенных сюда из матрикса.
Все переносчики встроены во внутреннюю мембрану митохондрий, кроме никотинамидных дегидрогенказ. Они составляют дыхательный ансамбль, тысячи таких ансамблей существуют в митохондрии и потребляют 90-95% кислорода, который используется клеткой. Два атома водорода отнимаются от субстрата и передаются на О2 с образованием Н2О. Разность потенциалов на двух концах полной цепи составляет 1.14V.
Митохондриальное окисление. НИКОТИНАМИДНЫЕ ДЕГИДРОГЕНАЗЫ (НАДГ)
Небелковая часть этих ферментов представляет собой динуклеотид: НИКОТИНАМИД-АДЕНИНДИНУКЛЕОТИД (НАД+) или НИКОТИНАМИДАДЕНИНДИНУКЛЕОТИДФОСФАТ (НАДФ+).

Студенты обязаны знать формулу НАД(Ф) и механизм присоединения к нему водорода. НАД(Ф) содержит производное витамина РР — никотинамид. (см. раздел «Витамины»).
НАД+ и НАДФ+ входят в состав каталитического центра НАДГ. Они являются КОФЕРМЕНТАМИ, так как связаны с белковой частью слабыми типами связей — могут легко диссоциировать. Они присоединяются к белковой части только в момент протекания реакции. Реакция, которую катализируют НАДГ — это реакция окисления субстрата.

Известно около 150 НАДГ, которые различаются по строению белковой части (апофермента).
Апоферменты большей части НАДГ способны присоединять или только НАД, или только НАДФ, и лишь немногие способны соединяться и с тем, и с другим коферментами. НАДГ, участвующие в митохондриальном окислении, находятся в матриксе митохондрий, в отличие от большинства других участников дыхательной цепи, которые встроены во внутреннюю мембрану. НАДГ можно встретить и в цитоплазме клеток. Мембрана митохондрий непроницаема для НАД(Ф), поэтому митохондриальный и цитоплазматический НАД(Ф) никогда не смешиваются. В митохондриях содержится очень много НАД и почти нет НАДФ, а в цитоплазме — наоборот — очень много НАДФ и почти нет НАД.
Из матрикса митохондриальный НАД×Н2 отдает два атома водорода на «комплекс I», встроенный во внутреннюю мембрану митохондрий.
Митохондриальное окисление. КОМПЛЕКС I
В составе комплекса находится 26 полипептидных цепей общей массой 800 кДа. Комплекс содержит следующие небелковые компоненты: Флавинмононуклеотид (ФМН), 5 центров FeS (железо-серные центры): FeS1a, FeS1b FeS2, FeS3, FeS4.
В транспорте водорода по дыхательной цепи в этом комплексе принимает участие ФМН.

Одновременно с протонами транспортируются и электроны. Наибольшие перепады редокс-потенциала наблюдаются между железо-серными белками, расположенными в следующем порядке:
ФМН → FeS1a → FeS1b → FeS3 → FeS4 → FeS2
Комплекс I – интегральный белковый комплекс. Используя энергию, выделяющуюся при переносе электронов по дыхательной цепи, он транспортирует 4 протона из матрикса в межмембранное пространство – комплекс I работает как протонный генератор. Точный механизм этого транспорта до сих пор неизвестен.
Далее комплекс I восстанавливает промежуточный переносчик KoQ (убихинон).

Это жирорастворимое низкомолекулярное вещество, содержащее длинную изопреновую цепь, не имеет белковой части. КоQ принимает водород от комплекса I. Образовавшийся КоQH2 отдает водород на комплекс III.
Митохондриальное окисление. КОМПЛЕКС III.
В своем составе содержит цитохромы – сложные белки, содержащие небелковый компонент — простетическую группу, сходню по строению с небелковой частью гемоглобина – гемом.
1) Цитохромы b, имеющие в своем составе два типа простетических групп тетрапиррольной структуры — «гем». Известно два гема цитохромов: be, обладающий низким окислительно-восстановительным потенциалом и bh с высоким окислительно-восстановительным потенциалом. Строение простетической группы цитохромов группы b, похожей на гем белка гемоглобина, представлено на рисунке. Его необходимо выучить.
 2)FeSIII – железо-серный кластер.
2)FeSIII – железо-серный кластер.
Цитохром С1. Имеет в своем составе особый гем типа «с».
Друг от друга цитохромы могут отличаться:
Строением белковой части;
Значением окислительно-восстановительного потенциала;
Строением радикалов, расположенных по периферии гема;
Присоединением гема к белковой части – в некоторых случаях гем присоединен к ней ковалентной связью за счет радикалов цистеина, что характерно для цитохромов c1 и c.
От двух атомов водорода, которые переносятся на комплекс III от KoQ, дальше по цепи транспортируются только электроны, два протона (H+)комплекс III выбрасывает в межмембранное пространство вместе с еще одной парой протонов, которые подхватываются комплексом из матрикса. Таким образом, комплекс III в сумме выбрасывает в межмембранное пространство 4 протона. Поэтому комплекс III, как и комплекс I, является протонным генератором, и целью его работы также является создание DmH+.
Митохондриальное окисление. КОМПЛЕКС IV.
Комплекс IV называется цитохромоксидазой. Он способен захватывать из матрикса 4 протона. Два из них он отправляет в межмембранное пространство, а остальные передает на образование воды.
Благодаря многоступенчатой передаче энергия в дыхательной цепи выделяется не мгновенно, а постепенно (маленькими порциями) при каждой реакции переноса. Эти порции энергии не одинаковы по величине. Их величина определяется разницей между ОВП двух соседних переносчиков. Если эта разница небольшая, то энергии выделяется мало — она рассеивается в виде тепла. Но на нескольких стадиях ее достаточно, чтобы синтезировать макроэргические связи в молекуле АТФ. Такими стадиями являются:
| Стадии | Разность потенциалов |
| НАД/ФАД | 0.25V |
| Цитохромы b/cc1 | 0.18V |
| aa3/O2 | 0.53V |
Значит, на каждую пару атомов водорода, отнятых от субстрата, возможен синтез 3-х молекул АТФ.
 АДФ + Ф + ЭНЕРГИЯ → АТФ + Н2О
АДФ + Ф + ЭНЕРГИЯ → АТФ + Н2О
Макроэргическая связь — это такая ковалентная связь, при гидролизе которой выделяется не менее 30 кДж/моль энергии. Эта связь обозначается знаком ~.
Синтез АТФ за счет энергии, которая выделяется в системе МтО, называется ОКИСЛИТЕЛЬНЫМ ФОСФОРИЛИРОВАНИЕМ. Основная роль АТФ — обеспечение энергией процесса синтеза АТФ.
Для оценки эффективности работы системы МтО при окислении вычисляют КОЭФФИЦИЕНТ P/O. Он показывает, сколько молекул неорганического фосфата присоединилось к АДФ в расчете на один атом кислорода.
Для главной (полная) цепи Р/О=3 (10H+/2H+(затраты на освобождение АТФ из комплекса с ферментом) + 1H+ (затраты на транспорт фосфата)) = 3,3 (округляют до 3-х)), коэффициент полезного действия системы — 65%, для укороченной P/O=2 (6H+/2H+(затраты на освобождение АТФ из комплекса с ферментом) + 1H+ (затраты на транспорт фосфата)) = 2, для максимально укороченной P/O=1 (4H+/2H+(затраты на освобождение АТФ из комплекса с ферментом) + 1H+ (затраты на транспорт фосфата)) = 1.
Система МтО потребляет 90% кислорода, поступающего в клетку. При этом в сутки образуется 62 килограмма АТФ. Но в клетках организма содержится всего 20-30 граммов АТФ. Поэтому молекула АТФ в сутки гидролизуется и снова синтезируется в среднем 2500 раз (средняя продолжительность жизни молекулы АТФ — полминуты).
Митохондриальное окисление. Автономная саморегуляция системы
Если клетка организма находится в условиях покоя, то АТФ мало используется и накапливается. Поэтому снижается концентрация АДФ и Ф. В этих условиях АТФ-синтетаза уже не получает из цитоплазмы достаточно фосфата и АДФ для синтеза АТФ. Её активность понижается, и скорость движения протонов из межмембранного пространства в матрикс по протонному каналу этого фермента тоже падает. Поэтому сохраняется высокий градиент концентраций протонов на внутренней мембране митохондрий. В этих условиях энергии переноса водорода по цепи митохондриального окисления уже не хватает для выталкивания Н+ из матрикса в межмембранное пространство. Перенос водорода по цепи МтО тормозится и прекращается окисление субстратов.
Метаболизм в клетке регулируется отношением АТФ/АДФ. Это отношение характеризует ЭНЕРГЕТИЧЕСКИЙ ЗАРЯД КЛЕТКИ.

В норме ЭЗК = 0.85-0.90. Может изменяться от 0 до 1. Высокий ЭЗК тормозит синтез АТФ, и активирует использование АТФ (АТФ → АДФ + Ф)
Митохондриальное окисление. Биологическая роль
Главная его функция — обеспечение организма запасами энергии в форме АТФ.
Именно митохондрии поставляют клетке большую часть необходимого ей АТФ.
В сутки синтезируется до 62 кг АТФ, хотя одновременно в организме никогда не бывает больше 30-40 граммов этого вещества. Т.е. наблюдается очень быстрое восстановление расходуемых молекул АТФ.
Митохондриальное окисление. Функции митохондрий
МИТОХОНДРИИ: ОСОБЕННОСТИ ХИМИЧЕСКОГО СОСТАВА, СТРОЕНИЯ.
Митохондрии — органеллы клеток. Они имеют 2 мембраны наружную гладкую и внутреннюю с многочисленными складками – кристами, внутреннее пространство митохондрий заполнено матриксом.

Наружная мембрана содержит много белка порина, образующего гидрофильные каналы, которые пропускаю через мембрану неорганические ионы, метаболиты и даже небольшие белки (меньше 10кДа). Также она содержит ферменты: элонгазы (удлиняют молекулы насыщенных жирных кислот), кинуренингидроксилазу, моноаминооксидазу (маркер) и др.
Межмембранное пространство митохондрий содержит аденилатциклазу, нуклеозиддифосфаткиназы.
Внутренняя мембрана высокоспецифична, состоит на 70% из белков, которые выполняют каталитическую (окислительное фосфорилирование) и транспортную функцию. Внутренняя мембрана содержит ферменты: а). цепи окислительного фосфорилирования (цитохромоксидаза – маркер) б). СДГ в). β-оксибутират ДГ; г). карнитинацилтрансферазу, д). карнитинацилтранслоказу.
Внутренняя мембрана на 30% состоит из фосфолипидов, из них 20% приходиться на кардиолипин, который делает мембрану непроницаемой для всех ионов.
Матрикс на 50% состоит из белка и содержит сотни различных ферментов. Это ферменты: а). ЦТК; б). β-окисления жирных кислот; в). аминотрансферазы АСТ, АЛТ; г). глутамат ДГ д). фосфоенолпируваткарбоксилазу е). пируват ДГ. Также матрикс содержит несколько копий митохондриальной ДНК, митохондриальные рибосомы и тРНК.
В клетке содержится от сотни до тысячи митохондрий, их размер 2-3 мкм в длину и 1 мкм в ширину.
Метаболические и гомеостатические функции митохондрий
В митохондриях происходит: синтез АТФ и теплопродукция в реакция окислительного фосфорилирования; β-окисления жирных кислот; реакции ЦТК, через ЦТК протекают некоторые реакции глюконеогенеза, переаминирования, дезаминирования, липогенеза и синтеза гема, осуществляется интеграция белкового, липидного и углеводного обмена.
Причины и последствия повреждений митохондрий
Повреждение внутренней мембраны митохондрий химическими и физическими факторами приводит разобщению окислительного фосфорилирования, нарушению синтеза АТФ, торможению анаболических реакций, межмембранного транспорта и всех видов обмена веществ.
Митохондриальное окисление.
Оксидазный путь использования кислорода в клетке
Оксидазный путь потребления кислорода протекает в митохондриях, потребляет 90% О2 и обеспечивает процесс окислительного фосфорилирования.
Окислительное фосфорилирование — синтез АТФ из АДФ и Н3РО4 за счет энергии движении электронов по дыхательной цепи.
Окислительное фосфорилирование является основным источником АТФ в аэробных клетках.
Хемиосмотическая теория Митчелла
Для объяснения механизма окислительного фосфорилирования в 1961 году Митчеллом была предложена хемиосмотическая теория, которая включала четыре независимых постулата, касавшиеся функции митохондрий:
- Внутренняя мембрана митохондрий непроницаема для всех ионов.
- Она содержит ряд белков-переносчиков, осуществляющих транспорт необходимых метаболитов и неорганических ионов.
- При прохождении электронов по дыхательной цепи внутренней мембраны происходит перемещение Н+ из матрикса в межмембранное пространство.
- При достаточно большом протонном градиенте протоны начинают «течь» через АТФ-синтетазу, что сопровождается синтезом АТФ.
Теория сопряжения окисления и фосфорилирования Питера Митчелла.

Известно, что через мембрану митохондрии могут свободно проникать только небольшие незаряженные молекулы, а также гидрофобные молекулы. Энергия, которая выделяется при переносе электронов по цепи МтО, приводит к переносу протонов (Н+) из матрикса митохондрии в межмембранное пространство. Поэтому на внутренней мембране митохондрий образуется градиент концентраций протонов: в межмембранном пространстве Н+ становится много, а в матриксе остается мало. Образуется разность потенциалов 0.14V — наружная часть мембраны заряжена положительно, а внутренняя — отрицательно. Накопившиеся в межмембранном пространстве Н+ стремятся выйти обратно в матрикс по градиенту их концентраций, но митохондриальная мембрана для них непроницаема. Единственный обратный путь в матрикс для протонов — через протонный канал фермента АТФ-синтетазы, которая встроена (built-in) во внутреннюю мембрану митохондрий. При движении протонов по этому каналу в матрикс их энергия используется АТФ-синтазой для синтеза АТФ. Синтезируется АТФ в матриксе митохондрий.
После синтеза АТФ переносится в цитоплазму путем облегчённой диффузии по градиенту концентраций, поскольку основные процессы, в которых АТФ потребляется, протекают в цитоплазме.
Как происходит транспорт АТФ из митохондрий в цитоплазму?
Для этого используется специфический для АТФ транспортный белок — АТФ/АДФ-транслоказа. Это интегральный белок, локализован во внутренней мембране митохондрий.

Во внутренней мембране митохондрий есть белок-переносчик — АТФ/АТФ-транслоказа, который имеет 2 центра связывания: со стороны матрикса для АТФ, снаружи — для АДФ. При изменении конформации АТФ/АДФ-транслоказы АДФ переносится в матрикс, а АТФ — в межмембранное пространство, а затем — в цитоплазму, где используется.
Для образования АТФ в матрикс всё время должен поступать неорганический фосфат (Ф). Для этого во внутренней мембране митохондрий есть транспортная система, которая обеспечивает перенос фосфата в матрикс сопряженно с переносом Н+. Это белок-переносчик, который имеет 2 центра связывания: для Ф и Н+. Ф и Н+ вместе переносятся из межмембранного пространства в матрикс.
Известны некоторые вещества, которые способны разобщать процессы окисления и фосфорилирования, приводя тем самым к уменьшению коэффициента р/о. К ним относятся йодсодержащие гормоны щитовидной железы (тироксин, трийодтиронин), а также некоторые ксенобиотики (например, 2,4-динитрофенол). Такие вещества известны под общим названием «РАЗОБЩАЮЩИЕ ЯДЫ». Как действуют вещества-разобщители окисления и фосфорилирования? Они могут образовывать собственные протонные каналы во внутренней мембране митохондрий. Поэтому часть протонов, вместо того, чтобы идти обратно в матрикс по протонному каналу АТФ-синтетазы, уходит туда по каналам веществ-разобщителей. В результате АТФ образуется меньше, и часть энергии выделяется в виде тепла.
Современные представления о механизме окислительного фосфорилирования
В настоящее время открыты все основные компоненты окислительного фосфорилирования, изучено их строение и свойства. Открыты основные принципы окислительного фосфорилирования, регуляция и механизмы некоторых стадий.
Митохондриальное окисление.
Транспорт веществ через мембрану митохондрий
1). Внутренняя мембрана митохондрии проницаема для незаряженных простых молекул О2, Н2О,
СО2, NH3, а также для монокарбоновых кислот. Эти вещества проходят мембрану самостоятельно по градиенту концентраций.
2). Для ионов мембрана не проницаема, через мембрану их переносят специальные насосы за счет энергии электрохимического потенциала. Только на транспорт АТФ и АДФ расходуется около четверти всей энергии электрохимического потенциала.
Симпортом с Н+ в матрикс перемещается ПВК и Са2+.
Антипортом перемещаются:

В результате обменных механизмов антипорта поддерживается осмотическое равновесие.
Митохондриальное окисление. Теплопродукция
30-35% свободной энергии рассеивается в виде теплоты и используется теплокровными животными на поддержание температуры тела. Кроме того, дополнительное образование теплоты может происходить при paзобщении дыхания и фосфорилирования в бурой жировой ткани. Она содержит много митохондрий с большим количеством дыхательных ферментов и разобщающего белка термогенина (РБ-1, около 10% всех белков). Разобщение окислительного фосфорилирования в бурой жировой ткани позволяет генерировать тепло для поддержания температуры тела у новорождённых, зимнеспящих животных и у всех млекопитающих в процессе адаптации к холоду.
У взрослых людей важную роль в термогенезе при переохлаждении играют жирные кислоты. Охлаждение стимулирует выделение норадреналина, который активирует липазу в жировой ткани и гидролиз ТГ. Образующиеся свободные жирные кислоты способны не только быть топливом дыхательной цепи, но и переносить протоны через мембрану в матрикс митохондрий разобщая дыхание и фосфорилирование. Обратно жирные кислоты в ионизированной форме возвращаются с помощью переносчиков.
Статья на конкурс «био/мол/текст»: «Только ленивый не занимался митохондриями!» — сказал один профессор. И действительно, митохондрии — очень популярный объект исследования, ведь в них происходит множество сложнейших биохимических и биофизических процессов, обусловливающих широкий набор функций данных органелл. Но, несмотря на активность исследователей, многие механизмы этих процессов и свойства отдельных компонентов митохондрий остаются загадкой. Это связано в первую очередь с отсутствием подходящих неинвазивных методик. В ходе междисциплинарного проекта, проводимого группой биофизики клетки Биологического ф-та МГУ и лабораторией неорганического материаловедения химического ф-та МГУ совместно с коллегами из Германии и Дании, удалось создать методику на основе спектроскопии гигантского комбинационного рассеяния для селективного исследования цитохрома с непосредственно в живых митохондриях. Статья опубликована в журнале Scientific reports.
О, эти спектры! О, эти пики!
В тайны молекул можем мы заглянуть.
Так и работаем мы в этом ЛИКе*,
И не напрасно избрали свой путь!
* — Лаборатория искусственного климата (ЛИК)
— прежнее название корпуса каф. Биофизики биофака МГУ.
Жалюзи опущены. Выключен свет. Мы погружены во мрак. Капля мутной жидкости падает на серебряную подложку. Вспышка зелёного света. 30 секунд. Спектр. Так начинается наш длинный день. А теперь обо всём по порядку.
Эти важные органеллы
Как вы уже (наверное) догадались, капля мутной жидкости — не что иное, как суспензия митохондрий. Митохондрии — это клеточные органеллы* размером около 1 микрона. Основная функция митохондрий — это окисление питательных веществ и производство молекул АТФ — универсального источника энергии для большинства биологических процессов в клетках. Кроме того, митохондрии участвуют в инициации апоптоза, старении клеток, продукции активных форм кислорода (АФК) [1], метаболизме лекарств, выработке тепла, запасают в себе ионы кальция, синтезируют некоторые гормоны и т.д. [2]. Митохондрии обладают столь широким спектром функций, что нарушение их работы является причиной многих заболеваний: различных видов миопатий, сердечно-сосудистых заболеваний, диабета и многих других [3]. Большинство из них связаны с мутациями в генах, кодирующих белки митохондрий**. Но может быть и наоборот: некоторые заболевания или неправильный образ жизни могут привести к нарушению работы митохондрий, например ожирение [4] или употребление наркотиков [5]. С другой стороны, митохондрии как преобразователи энергии и, фактически, белковый электропровод представляют большой интерес для физиков — биоников [6].
* — Органеллами митохондрии стали не сразу: согласно теории симбиогенеза, они появились в эукариотических клетках как бактерии-симбионты. И за миллиарды лет обросли множеством функций [7].
** — Т.к. митохондрии передаются по наследству от матери, то потомство получает все генетические нарушения, которые присутствовали в материнских митохондриях, но в перспективе эту проблему можно будет решить с помощью донорских митохондрий [8].
Митохондрии: как всё устроено
Митохондрии состоят из двух мембран: внешней, проницаемой для ионов и некоторых белков, и внутренней, ограничивающей внутреннее пространство митохондрий — матрикс. Пространство между мембранами так и называется — межмембранное пространство (ММП). Внутренняя мембрана образует множество инвагинаций (так называемых крист). В матриксе митохондрий расположены ферменты цикла Кребса, а во внутренней мембране митохондрий находится четыре белковых комплекса дыхательной цепи (электрон-транспортной цепи, ЭТЦ) и АТФ-синтаза (рис. 1). Также в митохондриях есть подвижные элементы дыхательной цепи: убихинон во внутренней мембране и цитохром с в ММП. Принцип работы дыхательной цепи — окисление субстратов, поступающих из цикла Кребса, и перенос электронов от этих субстратов по кофакторам ЭТЦ на конечный акцептор — кислород. Есть определенная закономерность в последовательности переноса электрона: электроны поступают от донора с более отрицательным редокс-потенциалом* (Еred/ox) к акцептору с более положительным Еred/ox. Таким образом, электрон как будто движется вниз по течению (рис. 2). Во время переноса электронов некоторые комплексы дыхательной цепи закачивают протоны из матрикса в ММП, тем самым создавая электрохимический потенциал на внутренней мембране митохондрий, который используется для синтеза АТФ (рис. 1) [2].
* — Окислительно-восстановительный потенциал — синоним редокс-потенциала (от англ. reduction — восстановление, oxidation — окисление).

Рисунок 1. Во внутренней мембране митохондрий локализованы комплексы дыхательной цепи: I — НАДН-дегидрогеназа, II — сукцинат-дегидрогеназа, III — цитохром b–c1-комплекс, или цитохром с-редуктаза, IV — цитохром а–а3, или цитохромоксидаза, и фермент АТФ-синтаза. Q — убихинон, «с» в зелёном кружке — цитохром с. Чёрными стрелками показан транспорт электрона, пунктирной стрелкой — диффузия цитохрома с от комплекса III к IV, зелеными стрелками — перенос протона.

Рисунок 2. Метафорическое изображение среднеточечных редокс-потенциалов различных переносчиков ЭТЦ митохондрий в энергетической шкале (вольт) на фоне реки Тохмайоки в Карелии.
Несмотря на то, что с митохондриями работали корифеи науки, такие как Кребс, Митчелл, Ленинджер, Чанс, Скулачёв и многие другие, благодаря которым мы столько знаем об этих органеллах, всё же многие митохондриальные процессы и свойства переносчиков электронов до сих пор не изучены. Это связано в первую очередь с отсутствием подходящих неинвазивных методик. В этом году нашей лабораторией в сотрудничестве с коллегами из других факультетов МГУ, а также из Дании и Германии, удалось создать методику для селективного исследования конформации гема цитохрома с непосредственно в живых митохондриях [9].
Что такое конформация гема и почему она важна?
Если приглядеться к комплексам дыхательной цепи (рис. 1), то в трёх из них можно увидеть цитохромы — гем-содержащие белки, которые являются кофакторами, участвующими в переносе электронов. А ещё один цитохром диффундирует в ММП. Всего в митохондриях имеется три типа цитохромов: а, b и с. Они очень похожи и отличаются только боковыми радикалами (рис. 3).

Рисунок 3. Гемы цитохромов разных типов. Каждый гем состоит из четырех пятичленных азотосодержащих циклов, связанных между собой. Это т.н. порфирины. Атомы азота координируют атом железа (Fe). Гемы отличаются боковыми группами, и только гем типа с связан с белком ковалентно. Рисунок из [2].
Во время переноса электрона меняется редокс-состояние гемового железа цитохромов Fe3+/Fe2+, что приводит к изменению конформации всего гема. Эти два параметра (Еred/ox и конформация) всегда связаны друг с другом. Кроме того, изменение в белковом окружении тоже приводит к изменению конформации гема [10, 11]. А так как Еred/ox определяет эффективность переноса электрона, то конформация гема — вещь важная. К тому же, чтобы произошел перенос электрона с одного цитохрома на другой, нужно, чтобы гемы этих цитохромов находились на определенном расстоянии друг от друга и были правильно ориентированы (рис. 4). Поэтому любые изменения в цитохромах могут отражаться на эффективности переноса электрона, т.е. на работе всей дыхательной цепи и, как следствие, энергообеспечении клеток [12, 13].

Рисунок 4. а. Расположение гемов цитохромов с и с1 при переносе электрона. б. Различные конформация гема цитохромов. Рисунки из [12] и [13].
Наиболее интересен в этом отношении цитохром с, так как это единственный мобильный переносчик электронов, курсирующий в ММП (все остальные цитохромы заякорены в комплексах ЭТЦ), поэтому он наиболее подвержен изменениям, которые могут происходить в митохондриях. Помимо этого выход цитохрома с запускает каскад реакций, ведущих к апоптозу, и в этом процессе тоже важен его редокс-потенциал [14].
Возникает вопрос, как понять, в какой конформации и в каком редокс-состоянии находится гем цитохрома с в митохондриях?
Как увидеть конформацию гема и оценить Еred/ox?

Рисунок 5. Сверху показан гем типа b, который содержится в эритроцитах, снизу — спектр КР эритроцита. Различные группы атомов и соответствующие им пики обозначены одним цветом. Рисунок из [20].
Численно можно определить Еred/ox с помощью электрохимических методов. Однако для этого нужные комплексы дыхательной цепи изолируют и помещают в совершенно иные условия среды, что может само по себе изменить их свойства. Благодаря таким мощным методам как ЯМР и рентгеноструктурный анализ, мы знаем, в какой конформации находятся белки и кофакторы дыхательной цепи в окисленной и восстановленной форме. Но нас интересует их состояние в максимально естественных условиях, а также соотношение этих состояний при работе дыхательной цепи. Есть несколько неинвазивных методов для определения окислительно-восстановительного состояния цитохромов. Например, абсорбционная спектроскопия. Спектр поглощения митохондрий — это суммарный спектр всех имеющихся цитохромов в митохондриях, а также ФАД. Но из-за того, что все типы цитохромов очень похожи и, соответственно, обладают схожими спектрами поглощения, а их окислительно-восстановительное состояние всё время меняется при нормальной работе дыхательной цепи, становится сложно оценить вклад различных цитохромов в спектр [15].
По спектрам флуоресценции НАДН и ФАД+ можно примерно оценить редокс-состояние только комплексов I и II. А по потенциалу на внутренней мембране митохондрий можно судить только о работе митохондрии в целом [16].
Напрямую получение информации о конформации и редокс-состоянии цитохрома с в интактной (целой и функционирующей) митохондрии — это крайне сложная задача, ведь его конформация постоянно меняется при работе ЭТЦ, он диффундирует и взаимодействует с мембранными комплексами. Наличие метода, который бы позволял это сделать, значительно расширило бы наши знания о влиянии цитохрома с на активность дыхательной цепи и его вкладе в развитие митохондриальных патологий.
К митохондриям от чистого сердца
Несколько лет назад в работах нашей группы вопрос о неинвазивном методе исследования цитохромов митохондрий в клетках уже поднимался. На тот момент сотрудники лаборатории исследовали влияние окислительного стресса на кардиомиоциты (клетки сердечной мышцы), методом комбинационного рассеяния света (КР, Raman spectroscopy) [17]. Подробно об этом рассказывается в статье «Спектроскопия КР: новые возможности старого метода» [18].
Спектр комбинационного рассеяния молекулы представляет собой колебательный спектр, пики которого характеризуют нормальные частоты колебаний молекулы. Отдельные группы атомов вносят свой вклад в различные колебания молекулы, поэтому можно сказать, что каждый пик на спектре КР характеризует колебания определённой группы атомов (рис. 5) [19, 20]. Таким образом, спектр КР — нечто вроде «отпечатков пальцев» молекулы. И так же, как по отпечаткам пальцев можно отличить родных братьев, по спектрам КР можно отличить схожие по строению молекулы. Кроме того, спектры КР чувствительны к изменениям конформации молекул, ведь если конформация меняется, то меняются и нормальные частоты колебаний.
В ходе работы с кардиомиоцитами было показано, что спектры КР разных типов цитохромов и миоглобина (который, как и гемоглобин, содержит гем типа b) можно различать по спектрам КР интактных клеток и даже определять их окислительно-восстановительное состояние. Позже Н.А. Браже с другими сотрудниками успешно провели подобное исследование на целом сердце, т.е. in situ (рис. 6) [21].

Рисунок 6. Регистрация спектра КР целого сердца. Рисунок из [21].
Однако у цитохромов есть одна особенность — спектр КР интенсивен только для восстановленных их форм, тогда как спектр окисленных состояний обладает очень низкой интенсивностью. Поэтому основным критерием определения восстановленности цитохромов является интенсивность сигнала. Но если мы ориентируемся только на интенсивность, то можем потерять важную информацию об изменениях в исследуемой системе! Ведь интенсивность зависит и от количества молекул, и от выраженности тех или иных колебаний, и от других параметров. К тому же, для определения точного положения пиков большинство исследователей, имеющих дело с цитохромами, вынуждены искусственно восстанавливать образцы, что заведомо нарушает их нативность.
Отчасти проблему решили японские учёные. Они определили, какие пики на спектре КР характерны только для восстановленных, а какие — только для окисленных цитохромов [22]. Однако проблема интенсивности спектров по-прежнему сохранялась. Нужно было её решать!
Усиливая сигнал
Один из способов усилить сигнал КР — это поместить исследуемую молекулу вблизи наночастицы благородного металла, например, золота или серебра. Эти металлы обладают свободными поверхностными электронами, а квант коллективных колебаний таких электронов называется плазмон. Если частота падающего света входит в резонанс с частотой колебаний поверхностных электронов металла, то возникает эффект плазмонного резонанса, который приводит к многократному усилению электромагнитного излучения вблизи наночастицы. Этот эффект используют в методе поверхностного плазмонного резонанса (surface plasmon resonance, SPR), в абсорбционной и флуоресцентной спектроскопии [23]. Но нас интересует способ усилить именно сигнал КР. И для этого существует спектроскопия гигантского комбинационного рассеяния (ГКР, англ. Surface enhanced Raman spectroscopy, SERS)*.
* — Подробности КР- и ГКР-спектроскопии даны в предшествующей статье: «Спектроскопия КР: новые возможности старого метода» [18].
Спектры КР и ГКР цитохрома с хорошо известны [24, 25]. И что самое замечательное, в отличие от спектров КР, спектры ГКР окисленного и восстановленного цитохрома с практически не отличаются по интенсивности. Так почему бы не зарегистрировать спектр ГКР митохондрий?
Идея пришла от О.В. Сосновцевой, профессора Копенгагенского университета, с которой наша лаборатория сотрудничает не первый год. Она даже предложила «подсобить» с митохондриями, так как с ними работали в соседней лаборатории, и они готовы были поделиться.
На тот момент было только две работы по изучению митохондрий методом ГКР и его разновидностью TERS (когда плазмонная частица находится на острие иглы кантилевера атомно-силового микроскопа, и сигнал ГКР регистрируют при сканировании поверхности образца). В первой работе [26] исследователи регистрировали спектр ГКР только от белков и липидов митохондрий, но не от цитохромов. А во второй работе [27] использовали не нативные, а высушенные митохондрии. Но как применить метод ГКР для изучения цитохромов живых митохондрий? Прежде чем ответить на этот вопрос, следует слегка переместиться в прошлое…
Междисциплинарность — залог успеха
В 2010 году, когда интерес к нанотехнологиям был на высоте, академик Юрий Дмитриевич Третьяков, в то время декан факультета наук о материалах (ФНМ) МГУ, решил начать межфакультетское сотрудничество с биологами для проведения совместных исследований методом ГКР. В итоге сотрудничество было установлено с лабораторией биофизики клетки под руководством Г.В. Максимова. Так наша лаборатория познакомилась с заместителем декана ФНМ химиком-материаловедом член-корр. Е.А. Гудилиным. Совместно с группой Гудилина начались активные исследования по применению ГКР в биологии.
Дело в том, что ГКР — метод непростой и требует исключительно междисциплинарного подхода. Каждый биообъект уникален, и чтобы регистрировать от него сигнал ГКР, нужно разрабатывать уникальные наноструктуры, которые бы усиливали сигнал от нужного объекта, не повреждали его и выдерживали «атаку» физиологических многокомпонентных буферов.
Большинство работ, связанных с ГКР, предполагают адсорбцию молекул на поверхности наноструктур, пусть даже введённых внутрь клетки [28]. Это связано с тем, что усиление сигнала КР наблюдается только на очень небольшом расстоянии от поверхности металла [29]. Однако такой подход не всегда удовлетворяет запросам биологов. В идеале усиление сигнала должно распространяться на достаточно большое расстояние, чтобы можно было работать с целыми клетками и органеллами, при этом не повреждая их, и желательно не вводить ничего внутрь. Для этой цели нужно разрабатывать специфический дизайн наноструктур и придумывать методы их синтеза, чем и занималась группа под руководством Гудилина.
Наконец, длительные и упорные попытки разработать такие структуры для исследования живых клеток увенчались успехом. В 2012-м году дебютировали наноструктурированные подложки для усиления сигнала от примембранного гемоглобина в интактных эритроцитах [20]. За счёт определённой морфологии наноструктур удалось получить усиление сигнала на расстоянии более 10 нм, т.е. больше толщины мембраны эритроцита.
Раз это удалось сделать для эритроцитов, то можно сделать и для митохондрий!
Долгожданный спектр ГКР митохондрий
Жалюзи опущены. Выключен свет. Комната погружена во мрак. Капля суспензии падает на серебряную подложку. Вспышка зелёного света. 30 секунд. Спектр. Тот самый долгожданный спектр ГКР от изолированных сердечных митохондрий был получен! Оставалось только понять, от каких именно структур в митохондриях исходит сигнал.
С учётом размеров компонентов митохондрий (рис. 7) и того, что усиление наблюдается на расстоянии нескольких нанометров от наноструктур, можно было предположить, что спектр ГКР митохондрий будет преимущественно спектром цитохрома с, так как этот цитохром наиболее близко подходит ко внешней мембране митохондрий и, соответственно, к наноструктурам. В то же время остальные цитохромы закреплены в комплексах внутренней мембраны.

Рисунок 7. Схема эксперимента: изолированные сердечные митохондрии помещают на серебряную наноструктурированную поверхность и облучают зелёным лазером. На рисунке показаны размеры мембраны, межмембранного пространства (ММП) и участков комплексов ЭТЦ. VDAC — потенциал зависимый анионный канал, FCCP — протонофор, разобщающий электронный транспорт и синтез АТФ, олигомицин — ингибитор АТФ-синтазы, с в синем кружке — цитохром с. Рисунок из [9].
Это утверждение также подтверждалось при моделировании эффектов усиления КР серебряными наноструктурами, которые были использованы в работе. Группа наших коллег из лазерного центра Ганновера показала, что сложная морфология подложки со множеством углублений, в которые могут попадать митохондрии, позволило получать усиление на большом расстоянии (более 10 нм). А иерархическое устройство самих наноструктур увеличивало число мест контакта с мембраной митохондрий и, следовательно, число молекул цитохрома с, от которых можно было зарегистрировать сигнал ГКР. Если использовать те же наночастицы серебра, просто присоединенные к плоской подложке, то особого усиления не произойдет, что и подтверждалось в эксперименте (рис. 8).

Рисунок 8. а—в — Изображения наноструктур, полученные на сканирующем электронном микроскопе. а — масштаб 10 мкм; б — 1 мкм, в — 0,2 мкм. На изображениях хорошо видна иерархическая морфология серебряных наноструктур. г—д — две модели, которые использовались для математического моделирования эффекта усиления электрического поля наноструктурами. г — в случае, когда митохондрии помещают на плоскую поверхность с наночастицами серебра, не происходит значительного усиления сигнала. д — при использовании структур, образующих полости, покрытые наночастицами серебра, число мест контакта между митохондрией и наночастицами увеличивается, а усиление электрического поля направлено перпендикулярно поверхности, т.е. внутрь митохондрии. За счёт этого достигается многократное усиление сигнала ГКР цитохрома с. Рисунок из [9] с изменениями.
И действительно, полученный спектр ГКР митохондрий соответствовал спектрам цитохрома с! При использовании зелёного лазера в качестве возбуждающего света можно регистрировать сигнал только от цитохромов b и с, но не от цитохрома а, что облегчает задачу расшифровки спектров. Несмотря на схожесть структуры цитохромов типа b и с, они имеют ключевые пики на спектре, благодаря которым их нельзя перепутать (рис. 9). Таким образом, используемые наноструктуры давали усиление на достаточно большом расстоянии, чтобы зарегистрировать спектры от цитохрома с, но недостаточно большом, для того чтобы увидеть пики цитохромов b. И это как раз то, что нужно! Благодаря методу ГКР теперь можно исследовать селективно редокс-состояние и конформацию именно цитохрома с в живых функционирующих митохондриях.

Рисунок 9. а — Спектры ГКР интактных митохондрий, помещенных на серебряную наноструктурированную поверхность. 1 — Спектр искусственно восстановленных цитохромов митохондрий; 2 — спектр активно функционирующих интактных митохондрий. б — Для сравнения приведен спектр ГКР эритроцитов, который соответствует спектру гема типа b, и спектр окисленного изолированного цитохрома с. Красным цветом отмечены пики, по которым можно чётко отличить спектр гема b и с. Рисунок из [9] с изменениями.
Как и ожидалось, спектры ГКР цитохрома с митохондрий оказались очень чувствительны к изменению его окислительно-восстановительного состояния. Для этого было исследовано два воздействия: внесение протонофора FCCP и ингибитора АТФ-синтазы олигомицина.
FCCP встраивается во внутреннюю мембрану митохондрий и начинает переносить протоны из ММП в матрикс, минуя протонные каналы АТФ-синтазы. В результате этого исчезает электрохимический потенциал и прекращается синтез АТФ. Это явление называют разобщением дыхания и фосфорилирования. В результате разобщения количество АТФ снижается, а АДФ увеличивается [30]. В этом случае возрастает количество поглощённого кислорода и скорость окисления субстратов, а, следовательно, и количество окисленных молекул цитохрома с, что очень хорошо видно на спектрах ГКР. И наоборот, добавление ингибитора синтеза АТФ — олигомицина, который приводит к увеличению электрохимического градиента на фоне снижения скорости дыхания, увеличивает количество восстановленных молекул цитохрома с, что также выражено на спектрах ГКР митохондрий.
Таким образом, спектры ГКР митохондрий, являясь спектрами исключительно цитохрома с, оказались чувствительны к изменениям его конформации и редокс-состояния в процессе работы митохондрий.
Схема всей работы в одном ролике
Итоги
Спектроскопия гигантского комбинационного рассеяния позволяет многократно усилить сигнал КР от молекул вблизи наночастиц металла. Однако для проведения успешных экспериментов необходимо учитывать свойства как биологического объекта, так и наноструктур. «Ключевым моментом нашего достижения стал междисциплинарный подход к работе, в которую были вовлечены биологи, химики и физики», — поясняет одна из главных авторов проекта к.б.н. Надежда Браже, ставшая осенью 2015 года лауреатом премии L’Oreal Unesco для женщин в науке. — «Результатом такого подхода стало создание уникальной методики селективного определения редокс-состояния и конформации цитохрома с в живых функционирующих митохондриях, помещенных на специальную наноструктурированную поверхность. Разработанная методика поможет восполнить пробелы наших знаний о свойствах и поведении переносчиков электрона в митохондриях, а также может быть использована для разработки диагностических тестов для раннего выявления патологий митохондрий, чем и планируется заниматься в ближайшее время».
- Активный кислород: друг или враг, или О пользе и вреде антиоксидантов;
- Nelson D.L. and Cox M.M. Lehninger Principles of Biochemistry (5th Edition). NY: W.H. Freemanand Company, 2008;
- Vafai S.B. and Mootha V.K. (2012). Mitochondrial disorders as windows into an ancient organelle. Nature. 491, 374—383;
- В ооцитах мышей с ожирением нарушается работа митохондрий;
- Пробило на хавчик;
- Moore L., Gust D., Moore T.A. (2007). Bio-inspired constructs for sustainable energy production and use. L’Actualité Chim. 308–309, 50–56;
- Как появились митохондрии (рассказ, похожий на сказку);
- Трое в лодке: о легализации замены митохондрий;
- Brazhe N.A., Evlyukhin A.B., Goodilin E.A., Semenova A.A., Novikov S.M., Bozhevolnyi S.I. et al. (2015). Probing cytochrome c in living mitochondria with surface-enhanced Raman spectroscopy. Sci. Rep. 5, 13793;
- Battistuzzi G., Borsari M., Cowan J.A., Ranieri A., Sola M. (2002). Control of cytochrome c redox potential : axial ligation and protein environment effects. J. Am. Chem. Soc. 19, 5315–5324;
- Dolla A., Blanchard L., Guerlesquin F., Bruschi M. (1994). The protein moiety modulates the redox potential in cytochromes c. Biochimie. 76, 471–479;
- Solmaz S.R.N. and Hunte C. (2008). Structure of complex III with bound cytochrome C in reduced state and definition of a minimal core interface for electron transfer. J. Biol. Chem. 283, 17542–17549;
- Ma J.G., Zhang J., Franco R., Jia S.L., Moura I., Moura J.J. et al. (1998). The structural origin of nonplanar heme distortions in tetraheme ferricytochromes c3. Biochemistry. 37, 12431–12442;
- Brown G.C. and Borutaite V. (2008). Regulation of apoptosis by the redox state of cytochrome c. Biochim. Biophys. Acta. 1777, 877–881;
- Chess D.J., Billings E., Covian R., Glancy B., French S., Taylor J. et al. (2013). Optical spectroscopy in turbid media using an integrating sphere: mitochondrial chromophore analysis during metabolic transitions. Anal. Biochem. 439, 161–172;
- Starkov A. and Fiskum G. (2003). Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 86, 1101–1107;
- Brazhe N.A., Treiman M., Brazhe A.R., Find N.L., Maksimov G.V., Sosnovtseva O.V. (2012). Mapping of redox state of mitochondrial cytochromes in live cardiomyocytes using Raman microspectroscopy. PLoS One. 7, e41990;
- биомолекула: «Спектроскопия КР: новые возможности старого метода»;
- Горелик В. (1997). Комбинационное рассеяние света. Соросовский образовательный журнал. 6, 91–96;
- Semenova А.А., Goodilin E.А., Brazhe N.А., Ivanov V.K., Baranchikov A.E., Lebedev V.A. et al. (2012). Planar SERS nanostructures with stochastic silver ring morphology for biosensor chips. J. Mater. Chem. 22, 24530;
- Brazhe N.A., Treiman M., Faricelli B., Vestergaard J.H., Sosnovtseva O. (2013). In situ Raman study of redox state changes of mitochondrial cytochromes in a perfused rat heart. PLoS One. 8, e70488;
- Kakita M., Kaliaperumal V., Hamaguchi H. (2012). Resonance Raman quantification of the redox state of cytochromes b and c in-vivo and in-vitro. J. Biophotonics. 5, 20–24;
- Миграция энергии плазмонного резонанса: вторая жизнь оптической спектроскопии;
- Hu S., Morris I.K., Singh J.P., Smith K.M., Spiro T.G. (1993). Complete assignment of cytochrome c resonance Raman spectra via enzymic reconstitution with isotopically labeled hemes. J. Am. Chem. Soc. 115, 12446–12458;
- Delfino I., Bizzarri A.R., Cannistraro S. (2005). Single-molecule detection of yeast cytochrome c by Surface-Enhanced Raman Spectroscopy. Biophys. Chem. 113, 41–51;
- Karataş O.F., Sezgin E., Aydin O., Culha M. (2009). Interaction of gold nanoparticles with mitochondria. Colloids Surf. B. Biointerfaces. 71, 315–318;
- Vitol E.A., Orynbayeva Z., Bouchard M.J., Azizkhan-Clifford J., Friedman G., Gogotsi Y. (2009). In situ intracellular spectroscopy with surface enhanced Raman spectroscopy (SERS)-enabled nanopipettes. ACS Nano. 3, 3529–3536;
- Kneipp J., Kneipp H., Wittig B., Kneipp K. (2010). Novel optical nanosensors for probing and imaging live cells. Nanomedicine. 6, 214–226;
- Moskovits M. (1978). Surface roughness and the enhanced intensity of Raman scattering by molecules adsorbed on metals. J. Chem. Phys. 69, 4159;
- Северин С. Е. Биохимия. Москва: ГЭОТАР-МЕД, 2003. — 779 с..
| Сокращения: |
| АОС − антиоксидантная система |
| ИП – ишемическое прекондиционирование |
| СС система – сердечно-сосудистая система |
| КМ – кардиомиоциты |
| митоKАТФ – АТФ-зависимые K+-каналы |
| MPT-пора – mitochondrial permeability transition pore |
| mtDNA – митохондриальная ДНК |
| NCX – Na+/Ca2+ обменник |
| HIF-1 – Hypoxia Inducible Factor-1 |
| HO-1 – гемоксигеназа-1 |
| Nrf2 – nuclear factor – erythroid-derived 2 – related factor 2 |
| RCK – регуляторы K+-проводимости |
| BKCa-каналы – Ca2+-активируемые K+-каналы высокой проводимости |
| ВММ – внутренняя мембрана митохондрий |
| МФК – митохондриальные ферментные комплексы |
| ПМ – плазматическая мембрана |
| СДГ − сукцинатдегидрогеназа |
При чрезмерной физической нагрузке и многих патологических состояниях в клетках нарушаются
физиологические и обменные процессы, обусловленные недостатком кислорода. В первую
очередь страдают метаболически активные ткани, функционирование которых зависит от
митохондрий, играющих ключевую роль в энергетическом метаболизме клеток. Дисфункция
митохондрий вызывает снижение работоспособности кардиомиоцитов, нейродегенеративные
заболевания и “митохондриальные болезни”.
После открытия в 1949 г. дыхания митохондрий эти клеточные структуры активно изучаются
во многих лабораториях мира. Однако, несмотря на многочисленные исследования митохондрий,
механизмы цитопротекции и особенно кардиопротекции, опосредованные митохондриями,
остаются малоизученными и далекими от полного понимания. Наблюдения последних лет
показали, что в кардиомиоцитах (КМ) митохондрии, помимо основной энергопродуцирующей
функции, также активно вовлечены во внутриклеточную сигнализацию и регуляцию специализированных
функций КМ. При этом важную роль в реализации внутриклеточной сигнализации и функций
самих митохондрий играют ионы Ca2+. Их дисбаланс в митохондриях КМ приводит к нарушению сердечной деятельности и развитию
сердечно-сосудистой (СС) патологии [1]. Ранее мы уже отмечали патогенетическое значение ионов Ca2+ в развитии постишемических повреждений миокарда и приводили примеры участия Ca2+ в процессах, влияющих на энергетику КМ и сократимость миокарда [2]. В экспериментах с использованием митохондрий, изолированных из сердца крыс, была
изучена регуляция ионами Ca2+ проницаемости внутренней мембраны митохондрий (ВММ) и установлено значение митохондриальных
Ca2+-активируемых неспецифических пор (MPT-пор – mitochondrial permeability transition
pore) в индукции апоптоза и некроза КМ [3–5]. Но если механизмы участия митохондрий в развитии нарушений функциональной и структурной
организации КМ достаточно хорошо освещены, то сведений о митохондриальных механизмах
кардиопротекции, включая Ca2+-зависимые, в литературе мало, что может свидетельствовать о недостаточной изученности
этой проблемы [1]. В настоящее время митохондрии рассматриваются в качестве объектов направленной
лекарственной терапии при многих заболеваниях, в том числе сердечно-сосудистых, и
с этой точки зрения задача суммировать знания, накопленные в данной области клеточной
биологии, представляется важной и своевременной.
ВЛИЯНИЕ ГИПОКСИИ
НА ЖИЗНЕДЕЯТЕЛЬНОСТЬ КАРДИОМИОЦИТОВ И КЛЮЧЕВЫЕ ФУНКЦИИ МИТОХОНДРИЙ
Гипоксия является важнейшим звеном многих патологических процессов и хронических заболеваний,
включая заболевания сердечно-сосудистой системы, среди которых наиболее распространенной
является ишемическая болезнь сердца.
Молекулярную основу патогенетических нарушений при гипоксии и ишемии составляют, главным
образом, процессы нарушения ключевых функций митохондрий − АТФ-продуцирующей и Ca2+-депонирующей, что приводит к энергетической дисфункции КМ, нарушению возбудимости
миокарда, а также невозможности осуществления желудочковыми КМ сократительной функции
[2, 6].
В исследованиях, проведенных на модели изолированных митохондрий, была установлена
следующая последовательность изменений в клетках в результате прекращения доступа
кислорода:
“0−5 мин: снижение уровня АТФ в клетке в 2−4 раза, несмотря на активацию гликолиза;
5−15 мин: повышение внутриклеточной концентрации Са2+. Активация гидролитических ферментов, в том числе митохондриальной фосфолипазы А2. Са2+-перегрузка митохондрий при сохранении ими всех основных функций (митохондрии еще
не повреждены);
15−30 мин: гидролиз митохондриальных фосфолипидов фосфолипазой А2 и нарушение барьерных свойств митохондриальной мембраны. Реоксигенация ткани на этой
стадии приводит к активному набуханию митохондрий. Дыхательный контроль в митохондриях
нарушен, окислительное фосфорилирование разобщено, способность митохондрий накапливать
ионы кальция снижена;
30−60 мин: частичное восстановление функций митохондрий, временное повышение дыхательного контроля,
способности накапливать кальций. Механизм компенсаторных процессов, приводящих к временному
улучшению функций митохондрий, неизвестен, но, очевидно, что он связан с функцией
клетки в целом, так как при анаэробной инкубации изолированных митохондрий это явление
не наблюдается;
60−90 мин: необратимое повреждение митохондрий и полная гибель клеток” (цитируется по: [7]).
Важно отметить, что последовательность изменений в клетке в результате аноксии одинакова
для самых различных тканей. Это показали опыты со срезами тканей, изолированными клетками
и изолированными митохондриями [7].
Сердце является самым энергоемким органом, при этом основным источником энергии, обеспечивающим
сократимость миокарда, является АТФ, аккумулируемая в процессе дыхания митохондрий
[8]. Для того чтобы обеспечить миокард достаточным количеством АТФ, необходимо огромное
количество функционально активных митохондрий, которые при нормоксии занимают около
одной трети от общего объема КМ и соответственно более половины объема миофибрилл.
При гипоксии количество митохондрий в КМ многократно возрастает [1]. Дисфункция митохондрий, вызванная развивающейся гипоксией или ишемией/реперфузией,
приводит к нарушению клеточного дыхания, следствием чего являются аритмии и снижение
сократительной способности миокарда [8].
В настоящее время влияние нарушения синтеза АТФ в митохондриях на функциональную активность
КМ изучено в наибольшей степени. Установлено, что при уменьшении содержания в клетке
АТФ на 10−20% снижается активность всех энергозависимых процессов на 70−80%. Угнетение
процессов энергообразования в митохондриях сопровождается ослаблением β-окисления
липидов, следствием чего являются нарушение липидного обмена в КМ и накопление ацил-CoA-тиоэфиров,
ацилкарнитинов, церамидов и триглицеридов, обладающих потенциальной цитотоксичностью.
При этом также происходит подавление анаболических процессов, нарушение работы ионных
насосов и соответственно ионного гомеостаза в клетке, приводящего к нарушению специализированных
клеточных функций и снижению жизнедеятельности КМ [1, 7–9]. Более подробно о влиянии гипоксии на энергозависимые процессы в КМ мы писали ранее
[2].
Другим важным фактором, приобретающим патогенетическое значение при развитии гипоксии,
является окислительный стресс, который индуцируется АФК при нарушении функционирования
дыхательной цепи митохондрий КМ. Известно, что в процессе функционирования дыхательной
цепи при нормоксии происходит образование незначительных количеств супероксидного
радикала, являющегося побочным продуктом дыхательных комплексов. В функционально полноценных
митохондриях действие митохондриальной антиоксидантной системы, включающей глутатион,
тиоредоксин-2, глутатион-пероксидазу, фосфолипид-гидропероксид-глутатион-пероксидазу
и Mn-супероксиддисмутазу, предотвращает повреждение митохондриальных структур активными
формами кислорода [9]. При нарушении переноса электронов между компонентами дыхательной цепи в условиях
окислительного стресса при гипоксии генерация митохондриями супероксидного радикала
существенно усиливается. Недостаточность функций митохондриальной антиоксидантной
системы в этом случае будет способствовать развитию окислительного стресса, активизации
самоподдерживающихся процессов перекисного окисления липидов, окислительным повреждениям
белков и нуклеиновых кислот. В конечном итоге эти события приводят к снижению функций
клетки, накоплению мутаций в митохондриальной и ядерной ДНК. В свою очередь повреждение
митохондриальных генов способствует нарушению процесса переноса электронов в дыхательной
цепи, результатом чего является дополнительное усиление продукции свободных радикалов
в митохондриях [9, 10].
На сегодняшний день показано, что образуемые митохондриями активные формы кислорода,
воздействуя на внутриклеточные сигнальные механизмы, могут инициировать развитие в
миокарде гипертрофических и фиброзных изменений. Избыточный уровень АФК может также
способствовать уменьшению сократительной активности КМ и повышать чувствительность
миофиламентов к Са2+ вследствие нарушения в КМ ионообменных процессов, за счет повреждения клеточных мембран
и ионных каналов [10].
В настоящее время известно, что митохондрии играют центральную роль в реализации программируемой
клеточной гибели КМ, к которой приводит, прежде всего, нерегулируемое повышение внутримитохондриальной
концентрации Са2+ [11, 12].
Са2+-перегрузка митохондрий. Многочисленными исследованиями было показано, что при гипоксии происходит Са2+-перегрузка митохондрий, имеющая ряд негативных последствий для КМ. В числе главных
следует назвать разобщение окислительного фосфорилирования, что снижает аэробный выход
АТФ и препятствует восстановлению адекватного энергообеспечения миокарда; нарушается
пассивный ионный транспорт в митохондрии; происходит набухание митохондрий и нарушение
целостности внутренней митохондриальной мембраны.
На модели изолированных митохондрий было установлено, что накопление Са2+ в матриксе митохондрий происходит в две фазы. Первая фаза − это энергозависимый вход
ионов Са2+ в матрикс митохондрий, стимуляция дыхания и выброс протонов из матрикса. Во второй
фазе из-за открытия MPT-пор во ВММ накопленные ионы Са2+ начинают выходить из матрикса, и через некоторое время наблюдаются быстрое увеличение
проницаемости ВММ, обратный захват ионов водорода и высвобождение из матрикса ионов
K+. Развитие Са2+-перегрузки изолированных митохондрий усугубляется нарушением механизмов пассивного
ионного транспорта через наружную мембрану митохондрий [12].
Полагают, что в условиях ишемии и ишемии/реперфузии в митохондриях преобладают процессы
свободного окисления, способствующие накоплению Са2+ в матриксе митохондрий КМ. На это указывают прекращение захвата Са2+ митохондриями и даже выход из них уже накопленных ионов при подавлении процессов
свободного окисления. При этом повреждающее действие избыточного накопления ионов
Са2+ в КМ усиливается в условиях окислительного стресса, инициируемого гипоксией [10]. Моделирование ишемии и реперфузии в опытах на крысах одновременно со снижением
Са2+-связывающей способности митохондрий приводило к снижению трансмембранного потенциала,
уменьшению коэффициента АДФ/О (соотношение количественных величин добавленного АДФ
и поглощенного в течение состояния 3 кислорода) и замедлению дыхания митохондрий в
состоянии 3 [13].
В то же время не следует забывать о том, что митохондрии являются важными Ca2+-депонирующими органеллами КМ, которые по своей емкости лишь незначительно уступают
Са2+-емкости саркоплазматического ретикулума [3, 12]. Они принимают непосредственное участие в регуляции Са2+-гомеостаза в КМ и могут быть задействованы в механизмах адаптации КМ к окислительному
стрессу при реперфузии.
В целом к настоящему времени сложились представления о том, что митохондрии являются
первичными мишенями при гипоксии и ишемии/реперфузии, и процессы, происходящие в них,
могут приводить как к дисфункции и повреждению КМ, так и к их постишемической адаптации
[1, 6, 9, 13]. Поэтому многие исследования направлены на изучение роли митохондрий, митохондриальных
АФК и Са2+ в кардиопротекторном механизме ишемического и фармакологического прекондиционирования.
РОЛЬ МИТОХОНДРИАЛЬНОЙ ДЫХАТЕЛЬНОЙ ЦЕПИ В РАЗВИТИИ АДАПТАЦИИ К ГИПОКСИИ
Системный ответ организма на гипоксию включает различные адаптивные реакции (возрастание
объема альвеолярной вентиляции, повышение кислородной емкости крови, централизация
кровотока, увеличение сердечного выброса крови и др.). На клеточном уровне они направлены,
прежде всего, на сохранение энергосинтезирующей функции митохондрий. При этом используются
два типа механизмов: а) срочный компенсаторный, цель которого состоит в предотвращении
последствий острой гипоксии на молекулярном уровне и быстрое восстановление жизнедеятельности
клеток в постгипоксический период, и б) долгосрочные механизмы, которые формируются
в течение более длительного периода и способствуют увеличению неспецифической резистентности
клеток к дефициту кислорода. Эти механизмы базируются на регуляторном репрограммировании
активности митохондриальных ферментных комплексов (МФК) [14].
В нормоксических условиях работа дыхательной цепи митохондрий, как правило, зависит
от окисления НАД-зависимых субстратов − основного поставщика восстановительных эквивалентов
для дыхательной цепи через МФК I. Тем не менее 25−30% митохондриального дыхания в
этих условиях ассоциируются с МФК II [9, 14] и окислением сукцината, содержание которого в матриксе митохондрий невелико (0.2−0.4
ммоль/л) [15].
На ранней стадии гипоксии наблюдается активация электрон-транспортной функции МФК
I, способствующая усилению синтеза АТФ. Она отражает первичный компенсаторный механизм
мобилизации базовых энергетических ресурсов клетки в условиях сравнительно небольших
по силе внешних воздействий. При усиливающемся гипоксическом воздействии происходит
снижение редокс-потенциала первого комплекса и соответственно окисления НАД-зависимых
субстратов, а также компенсаторная активация альтернативных путей окисления ФАД-зависимых
субстратов, поставляющих восстановительные эквиваленты к МФК II− IV. Среди них особую
роль играет МФК II (сукцинатоксидазный путь окисления). В условиях гипоксии он имеет
термодинамические преимущества перед окислением НАД-зависимых субстратов цикла трикарбоновых
кислот, и, несмотря на то, что при этом сохраняются только два участка сопряжения
окисления и фосфорилирования, высокие скорости реакции обеспечивают достаточную энергетическую
эффективность процесса в целом [16]. Активация альтернативных метаболических путей, выполняющих функцию срочных компенсаторных
механизмов, позволяет сохранить поступление восстановительных эквивалентов на цитохромный
участок дыхательной цепи, благодаря чему электрон-транспортная функция МФК III и IV
и синтез АТФ в этом участке не нарушаются, что обеспечивает сохранение энергосинтезирующей
функции. Этот процесс направлен на использование энергетически более эффективного
в условиях гипоксии сукцинатоксидазного пути окисления дыхательных субстратов, благодаря
чему компенсируется снижение скорости окислительных превращений и синтеза АТФ, а также
устраняется характерный для гипоксии метаболический ацидоз и, как следствие, увеличивается
резистентность сердечной мышцы к дефициту кислорода [14, 16]. Более того, поскольку активация МФК II обусловливает приток Са2+ в митохондрии, увеличиваются его внутримитохондриальные запасы, реализуемые в условиях
гипоксии при снижении работоспособности миокарда [17].
Если перестройки работы субстратного участка дыхательной цепи при гипоксии не происходит,
то наблюдается резкая деэнергизация митохондрий (снижение мембранного потенциала,
потеря АТФ) и изменения в пуле адениннуклеотидов, нарушение дыхания, связанного с
окислением НАД-зависимых субстратов − донаторов электронов для комплекса I, поэтому
переключение путей окисления митохондриальных субстратов сопутствует практически любым
формам гипоксии или ишемии/реперфузии [14, 16, 18, 19].
По мнению Л.Д. Лукьяновой [9] репрограммирование дыхательной цепи митохондрий при гипоксии, приводящее к переключению
метаболических путей от окисления НАД-зависимых субстратов на окисление сукцината,
обусловлено следующими причинами:
1) чувствительностью сукцинатдегидрогеназы (МФК II) к редокс-состоянию пиридиннуклеотидов,
железо-серного кластера комплекса I и коэнзима Q, ее способностью активироваться при
росте степени их восстановленности при гипоксии и тормозиться при их окислении;
2) ограничением щавелевоуксусного торможения, подавляющего сукцинатзависимое дыхание,
так как в условиях высокой восстановленности дыхательной цепи оксалоацетат (конкурентный
ингибитор СДГ) восстанавливается в малат, и его ингибирующее действие на фермент уменьшается;
3) кинетическими преимуществами окисления сукцината − ФАД-зависимого субстрата − в
условиях высокой восстановленности НАДH (гипоксия) перед НАД-зависимыми субстратами
благодаря тому, что флавины в этих условиях сохраняются в более окисленном состоянии;
4) способностью сукцинат-зависимого окисления поддерживать высокую скорость доставки
восстановительных эквивалентов в дыхательную цепь и обеспечивать наибольший выход
богатых энергией соединений в единицу времени. Для биологической системы это важнее,
чем высокий коэффициент полезного действия, оцениваемый числом участков сопряжения
окисления и фосфорилирования, которое в данном случае уменьшается.
В процессе долгосрочной адаптации к гипоксии происходит транскрипционное ремоделирование
свойств основных субъединиц МФК I, в результате чего последний восстанавливает способность
к переносу электронов и окислительному фосфорилированию, утраченную в период срочной
адаптации. При этом постепенно снижается превалирующее влияние сукцинатоксидазного
окисления на энергообразование и дыхание митохондрий [14, 16, 19].
СИГНАЛЬНАЯ И КАРДИОЗАЩИТНАЯ РОЛЬ МАЛЫХ КОНЦЕНТРАЦИЙ МИТОХОНДРИАЛЬНЫХ АФК
Очевидно, что дисфункция митохондрий КМ при гипоксии находится в фокусе внимания современной
медицины. Однако, занимаясь изучением проблем фармакологической коррекции нарушений
КМ, вызванных гипоксическим или реперфузионным повреждением митохондрий, не следует
забывать, что эти органеллы высокоадаптированы к эндогенному окислительному стрессу,
вызываемому периодическим снижением уровня кислорода в крови и способны противостоять
физиологическим нагрузкам, сопряженным с кратковременным состоянием гипоксии [20]. При этом в КМ активируются механизмы антиоксидантной защиты и защиты митохондриальной
ДНК (mtDNA), усиливается дыхательный контроль митохондрий, в то же время увеличивается
продукция АФК, что способствует биогенезу митохондрий, повышению резистентности КМ
к экстремальным воздействиям и индукции системной метаболической кардиозащиты [1].
Гомеостаз АФК в клетке − необходимое требование нормального протекания многочисленных
физиологических и сигнальных процессов, в то время как избыточный внутриклеточный
уровень свободных радикалов кислорода приводит к окислительному стрессу, снижению
жизнеспособности клеток или даже их гибели [21–23].
При развитии гипоксии и ишемии/реперфузии при реоксигенезации миокарда продукция АФК
в КМ резко возрастает [23, 24]. Это связано с понижением концентрации АДФ и нарушением работы электрон-транспортных
цепей митохондрий, преимущественно на уровне комплексов I и III [25], снижением продукции митохондриальных антиоксидантов и внутримитохондриального пула
глутатиона [26], а также синтеза белка фратаксина, который участвует в регуляции митохондриального
транспорта железа [27] и ответственен за формирование митохондриального кластера Fe–S. Снижение синтеза
фратаксина приводит к увеличению внутриклеточного содержания Fe2+, в присутствии которого эндогенная перекись водорода может давать высокоактивный
гидроксильный радикал, обладающий наибольшей цитотоксичностью среди других АФК [28]. Способствует образованию гидроксильного радикала снижение парциального давления
кислорода в КМ во время ишемии, что также сопровождается переходом окисленных форм
железа Fe3+ в восстановленные Fe2+ [29].
Следует отметить, что во многих случаях АФК-индуцированной дисфункции КМ и эндотелиальных
клеток кровеносных сосудов СС системы сопутствует либо предшествует нарушение гомеостаза
Са2+ [30]. В КМ и эндотелиальных клетках кровеносных сосудов системы основным депо Са2+ служит сарко/эндоплазматический ретикулум, который может включать до 75% всех запасов
внутриклеточного Са2+, вторым по значению депо Са2+ являются митохондрии, которые вносят существенный вклад в агонист-индуцированную
мобилизацию Са2+ [31]. Ионы Ca2+ поступают в митохондрии, главным образом, через унипортер ВММ, а удаляются через
Na+/Ca2+ обменник (NCX) [31].
В эндотелиальных клетках сосудов митохондрии тесно контактируют с Са2+-каналами эндоплазматического ретикулума и плазматической мембраны (ПМ). Представляется
очевидным, что митохондрии эндотелия, в отличие от митохондрий КМ, не должны иметь
существенного значения для клеток, энергетические потребности которых примерно на
2/3 покрываются анаэробным гликолизом [32]. Однако результаты экспериментов свидетельствуют о том, что в клетках эндотелия
митохондрии, также как и в КМ, выполняют важные регуляторные и сигнальные функции,
осуществляющиеся с участием АФК, которые нарушаются при развитии патологии СС системы
и хирургических вмешательствах. Наиболее выраженные патологические изменения в клетках
СС системы, инициируемые АФК, возникают при гипоксии, а в случае ишемии − при реперфузии.
Основные источники АФК при реперфузии сосудов − NADPH-оксидазы, ксантиноксидаза и
дыхательная цепь самих митохондрий (МФК IV) [33]. Усиление свободнорадикальных процессов в митохондриях эндотелиальных клеток может
приводить к патологическому делению митохондрий и к продолжительному повышению внутримитохондриального
уровня ионов Са2+, сопряженному с ингибированием митохондриального обменника NCX активными формами
кислорода и со структурно-функциональным разобщением митохондрий и эндоплазматического
ретикулума [34, 35].
Роль митохондриального кальция в повреждении эндотелиальных клеток при реперфузии,
как и в целом, взаимосвязь кальция и АФК в клетках эндотелия, представляет собой важную,
но малоизученную проблему [29]. Имеющиеся данные позволяют предполагать, что при реоксигенации возникают инициируемые
АФК осцилляции цитозольного кальция [36], которые, в свою очередь, влияют на состояние митохондрий, усиливая вторичную генерацию
АФК [37] и экзоцитоз адгезивных молекул, усугубляющих дисфункцию эндотелия за счет инфильтрации
лейкоцитов [38].
Негативное влияние АФК на эндотелий сосудов во многом обусловлено повреждающим действием
радикалов кислорода на клеточные мембраны. При этом АФК могут активировать ряд каналов
эндоплазматического ретикулума и ПМ, включая IP3— и рианодинчувствительные каналы, а также некоторые катионные каналы суперсемейства
TRP для неуправляемого входа Са2+, что может вызвать кальциевую перегрузку клеток, увеличить проницаемость ВММ и стать
причиной программируемой гибели эндотелиальных клеток [39–43].
Однако, как уже неоднократно отмечалось, в сердечно-сосудистой системе АФК выступают
не только в качестве повреждающего фактора при чрезмерной интенсификации свободнорадикального
окисления, но выполняют защитную роль, выступая и в качестве индукторов редокс-чувствительных
кардиопротекторных сигнальных путей, ключевыми компонентами которых являются транскрипционные
факторы HIF-1 (Hypoxia Inducible Factor-1), и Nrf2 (nuclear factor – erythroid-derived
2 – related factor 2). О структуре и функциях HIF-1, как сигнальной молекулы, при
адаптации к гипоксии мы писали ранее [20]. В данной статье представляется важным рассмотреть компоненты другого, менее изученного
Nrf2-зависимого − сигнального пути, который активируется в условиях гипоксии и ишемии/реперфузии
и направлен на защиту клеток от повреждений, сопряженных с гипоксией [44].
Транскрипционный фактор Nrf2 относится к семейству ДНК-связывающих белков, содержащих
лейциновый мотив, так называемую “лейциновую молнию» (basic leucine zipper transcription
factor), и является основным молекулярным регулятором во внутриклеточной антиоксидантной
защите. Известно, что Nrf2 транслоцируется в ядро при возникновении окислительного
или электрофильного стресса [44]. В ядре Nrf2 связывается с регуляторами антиоксидантного ответа в промоторах многочисленных
генов, ответственных за синтез ферментов антиоксидантной защиты и детоксикации, таких
как супероксиддисмутаза-1, глутатионтрансфераза, глутаматцистеинлигаза, гемоксигеназа-1
(HO−1) и др. [45]. В ряде работ было показано, что сверхэкспрессия Nrf2 оказывает цитопротекторное
действие на клетки, в то время как отсутствие его экспрессии, напротив, увеличивает
чувствительность тканей к окислительному стрессу [45–50]. Несмотря на то что многие виды структурных нарушений клеток и тканей, вызванных
окислительным стрессом, связаны с дисфункцией митохондрий, до последнего времени не
было известно, защищает ли Nrf2 митохондрии от повреждений, индуцируемых АФК. В связи
с этим большой интерес представляют работы, в которых обнаружено влияние Nrf2 на биогенез
и функции митохондрий КМ в условиях окислительного стресса. В этих работах было показано,
что сверхэкспрессия Nrf2 предотвращает развитие индуцируемых АФК нарушений в митохондриях,
таких как повреждение митохондриальных сетей, потеря митохондриального мембранного
потенциала, снижение экспрессии цитохрома с. Обнаружение локализации Nrf2 в наружной мембране митохондрий предполагает прямое
взаимодействие Nrf2 со структурными компонентами митохондрий для сохранения их целостности
и функциональной активности [44, 51, 52].
В экспериментах in vitro и in vivo на крысах и кроликах изучалось действие Nrf2 на процессы митофагии и клеточной гибели,
инициирумой окислительным стрессом, как при остром повреждении миокарда, так и при
развитии сердечной недостаточности [44, 53–57]. Было установлено, что Nrf2 защищает от деструкции мтДНК и белки, участвующие в
биогенезе митохондрий, такие как Drp1, Drp2, hFis-1, mitofusin 1, mitofusin 2, OPA1
[44, 53, 57], а также подавляет протеотоксический некроз КМ путем усиления аутофагического клиренса
токсичных убиквитированных белков [58]. Установлено, что экспрессия Nrf2 в КМ снижает степень дезадаптивного ремоделирования
левого желудочка в ответ на перегрузку кровяным давлением. При этом дефицит Nrf2 на
системном уровне сопровождался ранним началом ремоделирования миокарда и последующим
развитием сердечной недостаточности [59, 60]. Следует отметить, что к сердечной недостаточности и другим нарушениям в СС системе
также могли приводить мутации самого гена Nrf2 либо полиморфизм Nrf2—регулируемых генов детоксикации и АОС (его ослабленные полиморфные формы), обусловленные
длительным окислительным стрессом, что может служить еще одним доказательством важности
этого фактора транскрипции для обеспечения нормальной жизнедеятельности КМ и непосредственно
митохондрий в условиях гипоксии и ишемии/реперфузии [61, 62].
В качестве сигнальной молекулы Nrf2 может участвовать в передаче сигналов по редокс-зависимым
сигнальным путям, в первую очередь, Akt/PKB/mTOR (mammalian target rapamycin)-пути,
который играет ключевую роль в физиологии клеток сердечно-сосудистой системы в норме
и при патологии [63, 64]. Активация образуемых mTOR-комплексов имеет важное значение для выживаемости клеток
миокарда при ишемии. В частности, получены данные, согласно которым фосфорилированые
mTOR-комплексы, главным образом mTORC1, проявляют кардиопротекторые свойства за счет
угнетения аутофагии и активации восстановительных процессов ишемизированного миокарда
[64].
РОЛЬ МИТОХОНДРИАЛЬНЫХ КАЛИЕВЫХ КАНАЛОВ В РЕАЛИЗАЦИИ КАРДИОЗАЩИТНЫХ ЭФФЕКТОВ ИШЕМИЧЕСКОГО
ПРЕКОНДИЦИОНИРОВАНИЯ
Учитывая важную роль калиевого транспорта в обеспечении нормального функционирования
митохондрий КМ, очевидно, что разработка мер по оптимизации внутримитохондриального
калиевого транспорта в условиях ишемии и ишемии/реперфузии является важным стратегическим
направлением в метаболической кардиопротекции и молекулярной терапии. В нормальной
физиологии КМ транспорт K+ в митохондриях имеет большое функциональное значение [65], но его роль многократно возрастает в условиях гипоксии, когда меняется клеточный
метаболизм КМ и активируются механизмы срочной клеточной адаптации.
В митохондриях различают несколько систем транспорта K+, включая пассивный транспорт, хотя с учетом большого электрохимического потенциала
(~−200 мВ с матриксной стороны митохондрий) скорость такой диффузии будет невелика
[66]. Что касается специфических систем транспорта, то в митохондриях существует система
электрогенного входа калия через специфические K+‑каналы и К+/Н+-антипортер [65, 67, 68], из которых K+-каналы рассматриваются не только в качестве объектов транспортной системы митохондрий,
но и c точки зрения прикладной медицины – в качестве основных объектов молекулярной
терапии и профилактики последствий ишемии и, главным образом, реперфузии, возникающей
при возобновлении кровотока в зоне ишемического повреждения миокарда [65]. При этом подразумеваются, прежде всего митохондриальные АТФ-зависимые K+-каналы (митоKАТФ), интерес к которым был стимулирован открытием кардиозащитного действия прерывистой
гипоксии (феномен, получивший название ишемического прекондиционирования) [69].
Ишемическое прекондиционирование (ИП) − это адаптация миокарда к ишемии, развивающаяся
после нескольких повторяющихся кратковременных эпизодов ишемии/реперфузии и приводящая
к возникновению устойчивости сердечной мышцы к последующей более длительной ишемической
атаке. Согласно данным литературы, ишемическое прекондиционирование позволяет в некоторых
случаях предотвратить развитие инфаркта миокарда, а при его возникновении обеспечивает
меньшие размеры зоны некроза, уменьшает вероятность появления аритмий, в том числе
и реперфузионных, предупреждает развитие дисфункции левого желудочка [2].
Исследования, выполненные к настоящему времени, показывают, что механизм ИП представляет
собой каскад метаболических и сигнальных процессов, начинающийся с активации рецепторов
ишемическим стимулом (триггером), последовательным усилением сигнала и его передачи
на конечные эффекторные мишени, включая сарколеммальные и митохондриальные АТФ-зависимые
калиевые каналы [70].
При исследовании молекулярных механизмов ИП было установлено, что KАТФ-каналы, открывающиеся в процессе ишемии в сарколемме и митохондриях КМ, играют центральную
роль в реализации защитных эффектов прекондиционирования. Это доказывается тем, что
кардиопротекторный эффект ИП может быть полностью блокирован ингибиторами KATФ-каналов типа глибенкламида и 5‑гидроксидеканоата [71]. Первоначально ответственными за ИП считались исключительно сарколеммальные KATФ-каналы, однако позднее стало очевидным, что важнейшую роль в развитии этого процесса
играют митоKATФ [65, 72]. KATФ-каналы в митохондриях специфически открываются пинацидилом и диазоксидом, а сарколеммальные
– только пинацидилом, и блокируются 5-гидроксидеканоатом и глибенкламидом, однако,
в отличие от последних, ингибирование митоKATФ происходит при значительно более низких концентрациях этих веществ [65]. Подробнее о роли митоKATФ-каналов в процессе ИП, их структуре и регуляции мы писали ранее [2].
В данной статье мы хотим привлечь внимание к Ca2+-активируемым K+-каналам высокой проводимости (BKCa) − другой малоизученной разновидности митохондриальных K+-каналов, принимающих участие в механизме кардиопротекции [73].
Первоначально была изучена структура BKCa-каналов, локализованных на плазматической мембране. Было установлено, что эти каналы
характеризуются большой проводимостью, а также чувствительностью к изменениям мембранного
потенциала и концентрации внутриклеточного Ca2+ [74]. Исследование структурно-функциональной организации каналов показало, что канальный
белок, кодируемой геном Kcnma1, образован четырьмя α-субъединицами, состоящими из N-внеклеточной терминали, 7 трансмембранных
доменов (S0−S7) и внутриклеточной C-терминали. Домен, являющийся сенсором потенциала,
включает сегменты S0−S4, а пора и селективный фильтр формируются линкером S5−S6 [75, 76].
Внутриклеточная C-терминаль, являющаяся самым протяженным доменом BKCa-канала, содержит две Ca2+-чувствительные области, известные как регуляторы K+-проводимости (RCK) 1 и 2. Исследования мутагенеза показали, что RCK1 содержит два
важнейших для Ca2+-регуляции остатка аспартата (D362/D367), а RCK2 − 5 консекутивных аспартатов в Ca2+-связывающих участках (“calcium bowl”), которые в сочетании друг с другом являются
достаточными для активации BKCa-канала при физиологических концентрациях Са2+ [77, 78]. Кроме Ca2+, BKCa-каналы могут активироваться миллимолярными концентрациями Mg2+. Эта активность связана с наличием Mg2+-сенсора, представляющего собой совокупность аминокислотных остатков (D99, N172 и
E374, E399), локализованных на разных α-субъединицах канала, но связанных электростатическими
взаимодействиями [79].
Важно отметить, что ген Kcnma1 при транскрибировании может подвергаться обширному альтернативному сплайсингу, что
приводит к появлению нескольких изоформ BKCa-каналов с различными функциональными характеристиками, включая потенциал- и Са2+-чувствительность, ответы на фосфорилирование и модуляцию арахидоновой кислотой, а
также субклеточную локализацию, в том числе митохондриальный таргетинг [80].
Первые доказательства того, что BKCa-каналы с проводимостью около 300 pS (в 150 мМ KCl) присутствуют на ВММ, были получены
в конце 90-х годов [81]. Канал был охарактеризован авторами с использованием митопластов − митохондрий,
лишенных внешней мембраны, выделенных из клеток глиомы LN-229. В дальнейшем BKCa-каналы митохондрий (митoBKCa) со сходными проводимостями (в диапазоне от 200 до 307 pS), были обнаружены и в других
клеточных системах.
К настоящему времени известно, что митoBKCa и порообразующие α-субъединицы BKCa-каналов ПМ кодируются одним и тем же геном (Kcnma1) [82]. Это объясняет, почему они имеют общие базовые биофизические свойства, включающие
большую проводимость и сходные ответы на изменение потенциала и [Ca2+]i, хотя, очевидно, что конкретные значения исследуемых параметров могут отличаться.
Сравнение свойств BKCa-каналов ПМ и митoBKCa в клетках глиомы человека (LN 229) показало, что проводимость первых составила 199
± 8 pS, а вторых – 278 ± 10 pS. Хотя обе разновидности каналов потенциал- и Ca2+-зависимы, их чувствительность была разной [83]. В конфигурации “inside-out” BKCa-каналы ПМ показали низкую чувствительность к потенциалу, что свидетельствует о незначительной
степени их открытия (Po) даже при высоком напряжении тока (Po < 0.1 при +80 mV and
~0.4 при100 mV) и аппликации 400 мкM Ca2+ с цитозольной стороны канала. В то время как порог открытия (Po) митoBKCa в конфигурации “on-mitoplast” и с той же концентрацией Ca2+ в среде достигала Po ~0.6 уже при отрицательном потенциале (−40 mV) [83]. В дальнейшем было показано, что клетки различных типов экспрессируют митoBKCa с различной потенциал- и Ca2+-чувствительностью.
Например, митоBKCa-каналы КМ морской свинки имели наиболее высокий Po ~ 0.9 при достаточно широком диапазоне
входного напряжения (от –60 до +60 мВ) и концентрации Са2+ [Са2+] 0.5 мкМ, из чего можно предположить, что их молекулярный состав (например, ассоциация
со вспомогательными субъединицами) может существенно отличаться от аналогов, экспрессируемых
в митохондриях глиомы, у которых при [Са2+] 1 мкМ, Po составляет 0.5 при входящем напряжении + 41 мВ (потенциал полуактивации,
V1/2 = = 41 мВ). Такая же изменчивость характерна и для BKCa-каналов плазматической мембраны, причем внутри одного и того же типа клеток [84]. Очевидно, что для того, чтобы понять природу изменчивости и гетерогенности BKCa-каналов, необходима их детальная биофизическая и молекулярная характеристика [80].
Интерес к митоBKCa-каналам, как уже отмечалось выше, преимущественно связан с открытием их защитной
роли при ишемии и реперфузии [73]. Авторам удалось показать, что фармакологическое прекондиционирование с применением
соединения NS1619-активатора BKCa-каналов, в концентрации 10–30 мкM до начала реперфузии приводило к улучшению диастолического наполнения левого желудочка и уменьшало зону
инфаркта. Оба эффекта отменялись при использовании 1 мкМ паксилина (рaxilline) – селективного
ингибитора BKCa. В пользу точки зрения, подразумевающей, что активаторы BKCa-каналов действуют именно на митохондриальные BKCa-каналы КМ, могут служить следующие аргументы: 1) NS1619 не может взаимодействовать
с BKCa плазматической мембраны, так как известно, что зрелые кардиомиоциты не экспрессируют
сарколеммальных BKCa [82, 85]; 2) поглощение K+ митохондриями ускоряется в присутствии NS1619 и замедляется в присутствии 100 нМ
ибериотоксина – селективного блокатора BKCa; 3) защитный эффект прекондиционирования с NS1619 не связан с миорелаксацией гладкомышечных
клеток сосудов, так как эффект миорелаксации, инициируемый активацией BKCa, является частью природного ионного механизма регуляции тонуса сосудов при гипоксии.
Дальнейшие исследования подтвердили высказанную точку зрения [80]. Более того, в ряде работ было доказано, что защитное действие прекондиционирования,
опосредованного активацией BKCa-каналов, связано с их ролью в поддержании нормального (физиологичного) уровня АФК,
препятствующего развитию окислительного стресса, и нарушению Ca2+-гомеостаза [82, 85]. Прямое измерение Ca2+-связывающей способности митохондрий КМ показало, что защитный эффект фармакологического
прекондиционирования, вызванный NS1619, отсутствовал у нокаутированных по Kcnma1 мышей [82]. В других экспериментах было показано, что применение ибериотоксина также приводило
к снижению Ca2+-связывающей способности митохондрий [87]. Эти результаты подразумевают, что активация BKCa-каналов в определенной степени может защитить митохондрии от неконтролируемого открытия
MPT-пор при гипоксии [87]. Механизм сопряжения BKCa-каналов с неселективными Ca2+-порами ВММ пока не совсем ясен, на митохондриях КМ такие исследования пока не проводились,
но, как показали исследования, проведенные на митохондриях из клеток мозга (близких
по интенсивности энергетического обмена и уровню потребления О2 к КМ), ингибирование mitoBKCa ибериотоксиом снижало количество Ca2+, необходимого для деполяризации митохондрий, и тем самым повышало степень открытия
MPT-пор [88]. При этом из митохондрий глиальных клеток увеличивался выход цитохрома с − маркера открытия MPT-пор, и апоптоза [87]. Предполагается, что ключевым белком, приводящим к взаимодействию митоBKCa-каналов и MPT-пор является проапоптотический белок Вах (Bcl-2 associated protein
X), который, непосредственно связываясь с mitoBKCa, и тем самым снижая их активность, инициирует открытие MPT-пор [87].
Подводя итог вышесказанному, можно с уверенностью утверждать, что митоBKCa-каналы играют важную роль в кардиопротекции и их, наряду с более изученными митоKАТФ-каналами, следует рассматривать в качестве перспективных молекулярных объектов кардиопротекции
и тангетной терапии ишемической болезни сердца.
Список литературы
-
Pohjoismäki J.L., Goffart S. The role of mitochondria in cardiac development and protection. Free Radic. Biol.
Med. 106: 345–354. 2017. https://doi.org/10.1016/j.freeradbiomed.2017.02.032 -
Shemarova I.V., Nesterov V.P., Korotkov S.M., Sobol’ K.V. Involvement of Ca2+ in the development of ischemic disorders of myocardial contractile
function. J. Evol. Biochem. Physiol. 53: 368−379. 2017. https://doi.org/10.1134/S0022093017050027 -
Han X., Xu J., Xu S., Sun Y., He M., Li X., Li X., Pi J., Yu R., Tian W. Role of mitochondrial permeability transition pore in mediating the inhibitory effect
of gastrodin on oxidative stress in cardiac myocytes in vitro. Nan Fang Yi Ke Da Xue
Xue Bao. 38: 1306−1311. 2018. https://doi.org/10.12122/j.issn.1673-4254.2018.11.05 -
Purroy R., Britti E., Delaspre F., Tamarit J., Ros J. Mitochondrial pore opening and loss of Ca2+ exchanger NCLX levels occur after frataxin depletion. Biochim. Biophys. Acta. 1864:
618−631. 2018. https://doi.org/10.1016/j.bbadis.2017.12.005 -
Wacquier B., Romero Campos H.E., González-Vélez V., Combettes L., Dupont G. Mitochondrial Ca2+ dynamics in cells and suspensions. FEBS J. 284: 4128−4142. 2017. https://doi.org/10.1111/febs.14296
-
Цыпленкова В.Г., Сутягин П.В., Суслов В.Б., Эттингер А.П. Особенности митохондриального аппарата кардиомиоцитов при различных заболеваниях
сердца и в эксперименте. Международный журнал прикладных и фундаментальных исследований.
8 (2): 53−56. 2014. [Cyplenkova V.G., Sutyagin P.V., Suslov V.B., E`ttinger A.P. Osobennosti mitoxondrial`nogo apparata kardiomiocitov pri razlichny`x zabolevaniyax
serdcza i v e`ksperimente. Mezh dunarodny`j zhurnal prikladny`x i fundamental`ny`x
issledovanij. 8(2): 53−56. 2014. (In Russ)]. -
Егорова М.В. Роль жирных кислот в адаптивных реакциях кардиомиоцитов при метаболической ишемии.
Дисс. докт. наук. Томск. 2014, 205 с. [Egorova M.V. Rol` zhirny`x kislot v adaptivny`x reakciyax kardiomiocitov pri metabolicheskoj ishemii.
Diss. dokt. nauk. Tomsk. 2014. (In Russ)]. -
Brown D.A., O’Rourke B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 88: 241−249. 2010. https://doi.org/10.1093/cvr/cvq231
-
Лукьянова Л.Д. Дизрегуляция аэробного энергетического обмена − типовой патологический процесс. В
кн.: Дизрегуляционная патология. М.: Медицина, С. 188− 215. 2002. [Luk`yanova L.D. Dizregulyaciya ae`robnogo e`nergeticheskogo obmena − tipovoj patologicheskij process.
V kn.: Dizregulyacionnaya patologiya. Moscow: Medicina. С. 188−215. 2002. (In Russ)]. -
Giordano F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 115:500−508.
2005. https://doi.org/10.1172/JCI200524408 -
Guimaraes C.A., Linden R. Programmed cell death: apoptosis and alternative deathstyles. Eur. J. Biochem. 217:
1638−1650. 2004. https://doi.org/10.1111/j.1432-1033.2004.04084.x -
Saris N.E., Carafoli E. A historical review of cellular calcium handling, with emphasis on mitochondria.
Biochemistry. 70: 187−194. 2005. https://doi.org/10.1007/s10541-005-0100-9 -
Лишманов Ю.Б., Нарыжная Н.В., Маслов Л.Н., Прокудина Е.С., Горбунов А.С., Цибульников
С.Ю. Функциональное состояние миокардиальных митохондрий при ишемии-реперфузии сердца
у крыс, адаптированных к гипоксии. Бюлл. эксп. биол. и мед. 15 (11): 589−592. 2013.
[Lishmanov Yu.B., Nary’zhnaya N.V., Maslov L.N., Prokudina E.S., Gorbunov A.S., Cibul’ni-kov
S.Yu. Funkcional’noe sostoyanie miokardial’nykh mitokhondrij pri ishemii-reperfuzii serdcza
u kryus, adaptirovannyux k gipoksii. Byull. eksp. biol. i med. 156 (11): 589−592.
2013. (In Russ).] -
Кирова Ю. И. Регуляторная роль сукцинат-зависимых сигнальных систем (HIF-1 и GPR91) при адаптации
к гипоксии. Автореф. докт. дисс. М., 2016. [Kirova Yu. I. Regulyatornaya rol’ sukcinat-zavisimyux signal’nyux sistem (HIF-1 i GPR91) pri adaptacii
k gipoksii. Avtoref. dokt. diss. Moscow, 2016. (In Russ)]. -
Komaromy-Hiller G., Sundquist P.D., Jacobsen L.J., Nuttall K.L. Serum succinate by capillary zone electrophoresis: marker candidate for hypoxia.
Ann. Clin. Lab. Sci. 27: 163–168. 1997. -
Лукьянова Л.Д. Биоэнергетическая гипоксия: понятие, механизмы, коррекция. Бюл. эксп. биол. и мед.
124 (9): 244−254. 1997. [Luk`yanova L.D. Bioe`nergeticheskaya gipoksiya: ponyatie, mexanizmy`, korrekciya. Byul. e`ksp. biol.
i med. 124 (9): 244−254. 1997. (In Russ)]. -
Levrat C., Louisot P. Study of the succinate dehydrogenase activation in permeabilized mitochondria through
the Ca2+-stimulated phospholipase A2. Biochem. Mol. Biol. Int. 34: 569−578. 1994. -
Дудченко А.М., Лукьянова Л.Д. Триггерная роль энергетического обмена в каскаде функционально-метаболических нарушений
при гипоксии. Проблемы гипоксии: молекулярные, физиологические и клинические аспекты.
Москва.: Истоки, 2004. С. 51−83. [Dudchenko A.M., Luk’yanova L.D. Triggernaya rol` e`nergeticheskogo obmena v kaskade funkcional no-metabolicheskix
narushenij pri gipoksii. Problemy` gipoksii: molekulyarny`e, fiziologicheskie i klinicheskie
aspekty`. Moskva.: Istoki, 2004. S. 51−83. (In Russ).] -
Кондрашова М.Н. Трансаминазный цикл окисления субстратов в клетке как механизм адаптации к гипоксии.
Фармакологическая коррекция гипоксических состояний. Москва: ВИНИТИ, 1989. С. 51−66.
[Kondrashova M.N. Transaminazny`j cikl okisleniya substratov v kletke kak mexanizm adaptacii k gipoksii.
Farmakologicheskaya korrekciya gipoksicheskix sostoyanij. Moskva: VINITI, 1989. S.
51−66. (In Russ)]. -
Shemarova I.V., Nesterov V.l. P., Korotkov S.M., Sylkin Yu.A. Evolutionary Aspects of Cardioprotection. J. Evol. Biochem. Physiol. 54(1): 8-21.
2018. https://doi.org/10.1134/S0022093018010027 -
Гамалей И.А. Роль активных форм кислорода в регуляции функций клетки. Дисс. докт. наук. Санкт-Петербург,
1999. 41 с. [Gamalej I.A. Rol` aktivny`x form kisloroda v regulyacii funkcij kletki. Diss. dokt. nauk. Sankt-Peterburg,
1999. 41 s. (In Russ)]. -
Dröge W. Free radicals in the physiological control of cell function. Physiol. Rev. 82: 47−95.
2002. https://doi.org/10.1152/physrev.00018.2001 -
Baines C.P. How and when do myocytes die during ischemia and reperfusion: the late phase. J.
Cardiovasc. Pharmacol. Ther. 16: 239−243. 2011. https://doi.org/10.1177/1074248411407769 -
Гребенчиков О.А., Лихванцев В.В., Плотников Е.Ю. Молекулярные механизмы развития и адресная терапия синдром ишемии-реперфузии. Анест.
и реаним. 3: 59−67. 2014. [Grebenchikov O.A., Lixvancev V.V., Plotnikov E.Yu. Molekulyarnye mexanizmy razvitiya i adresnaya terapiya sindrom ishemii-reperfuzii.
Anest. i reanim. 3: 59−67. 2014. (In Russ)]. -
Herrero A., Baria G. Localization of the site of oxygen radical generation inside the mitochondria. J.
Bioenerg. Biomembr. 32: 609−615. 2000. -
Кулинский В.И., Колесниченко Л.С. Обмен глутатиона. Успехи биол. химии. 31:157−179. 1990. [Kulinskij V.I., Kolesnichenko L.S. Obmen glutationa. Uspexi biol. ximii. 31:157−179. 1990. (In Russ)].
-
Pandolfo M. Frataxin deficiency and mitochondrial dysfunction. Mitochondrion 2: 87−93. 2002.
https://doi.org/10.1023/A:1005626712319 -
Владимиров Ю.А. Свободные радикалы и антиоксиданты. Вестник РАМН. 7: 43−51. 1998. [Vladimirov Yu.A. Svobodnyue radikalyu i antioksidantyu. Vestnik RAMN. 7: 43−51. 1998. (In Russ)].
-
Надеев А.Д., Гончаров Н.В. Активные формы кислорода в клетках сердечно-сосудистой системы. Компл. пробл. серд.-сосудист.
забол. 4: 80−94. 2014. [Nadeev A.D., Goncharov N.V. Aktivny`e formy` kisloroda v kletkax serdechno-sosudistoj sistemy`. Kompl. probl.
serd.-sosudist. zabol. 4: 80−94. 2014. (In Russ)]. -
Dhalla N.S., Temsah R.M., Netticadan T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 18: 655−673. 2000.
https://doi.org/10.1097/00004872-200018060-00002 -
Malli R., Frieden M., Trenker M., Graier W.F. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J. Biol. Chem. 280: 12114−12122. 2005. https://doi.org/10.1074/jbc.M409353200
-
Culic O., Gruwel M.L., Schrader J. Energy turnover of vascular endothelial cell. Am. J. Physiol. 273: C205−C213. 1997.
https://doi.org/10.1152/ajpcell.1997.273.1.C205 -
Pearlstein D.P., Ali M.H., Mungai P.T., Hynes K.L., Gewertz B.L., Schumacker P.T. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia.
Arterioscler. Thromb. Vasc. Biol. 22: 566−573. 2002. https://doi.org/10.1161/01.ATV.0000012262.76205.6A -
Paltauf-Doburzynska J., Malli R., Graier W.F. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J. Cardiovasc. Pharmacol. 44: 423−436.
2004. https://doi.org/10.1097/01.fjc.0000139449.64337.1b -
Jornot L., Maechler P., Wollheim C.B., Junod A.F. Reactive oxygen metabolites increase mitochondrial calcium in endothelial cells:
implication of the Ca2+/Na+ exchanger. J. Cell Sci. 112:1013−1022. 1999. -
Hu Q., Ziegelstein R.C. Hypoxia/reoxygenation stimulates intracellular calcium oscillations in human aortic
endothelial cells. Circulation. 102(20):2541−2547. 2000. https://doi.org/10.1161/01.CIR.102.20.2541 -
Camello-Almaraz C., Gomez-Pinilla P.J., Pozo M.J., Camello P.J. Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. 291: 1082−1088. 2006. https://doi.org/10.1152/ajpcell.00217.2006
-
Ichimura H., Parthasarathi K., Quadri S., Issekutz A.C., Bhattacharya J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung
venular capillaries. J. Clin. Invest. 111: 691−699. 2003. https://doi.org/10.1172/JCI17271 -
Hecquet C.M., Malik A.B. Role of H2O2-activated TRPM2 calcium channel in oxidant-induced endothelial injury. Thromb. Haemost.
101: 619–625. 2009. https://doi.org/10.1160/TH08-10-0641 -
Hu Q., Zheng G., Zweier J.L., Deshpande S., Irani K., Ziegelstein R.C. NADPH oxidase activation increases the sensitivity of intracellular Ca2+ stores to inositol 1,4,5-trisphosphate in human endothelial cells. J. Biol. Chem.
275: 15749−15757. 2000. https://doi.org/10.1074/jbc.M000381200 -
Poteser M., Graziani A., Rosker C., Eder P., Derler I., Kahr H., Zhu M.X., Romanin
C., Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for
expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol.
Chem. 281: 13588−13595. 2006. https://doi.org/10.1074/jbc.M512205200 -
Stoyanovsky D., Murphy T., Anno P.R., Kim Y.M., Salama G. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium. 21:
19−29. 1997. https://doi.org/10.1016/S0143-4160(97)90093-2 -
Sun L, Yau H.Y., Wong W.Y., Li R.A., Huang Y., Yao X. Role of TRPM2 in H2O2-induced cell apoptosis in endothelial cells. PLoS One. 7: 43186. 2012. https://doi.org/10.1371/journal.pone.0043186
-
Strom J., Xu B., Tian X., Chen Q. M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 30: 66−80. 2016.
https://doi.org/10.1096/fj.14-268904 -
Narasimhan M., Rajasekaran N.S. Exercise Nrf2 and antioxidant signaling in cardiac aging. Front Physiol. 7: 241.
2016. https://doi.org/10.3389/fphys.2016.00241 -
Lee J.M., Calkins M.J., Chan K., Kan Y.W., Johnson J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection
against oxidative stress in primary cortical astrocytes using oligonucleotide microarray
analysis. J. Biol. Chem. 278:12029−12038. 2003. https://doi.org/10.1074/jbc.M211558200 -
Nguyen T., Nioi P., Pickett C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative
stress. J. Biol. Chem. 284: 13291−13295. 2009. https://doi.org/10.1074/jbc.R900010200 -
Li W., Kong A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 48: 91−104.
2009. https://doi.org/10.1002/mc.20465 -
Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev Pharmacol. Toxicol. 53:
401−426. 2013. https://doi.org/10.1146/annurev-pharmtox-011112-140320 -
Kensler T.W., Wakabayashi N., Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47: 89−116. 2007. https://doi.org/10.1146/annurev.pharmtox.46.120604.141046
-
Piantadosi C.A., Carraway M.S., Babiker A., Suliman H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional
control of nuclear respiratory factor-1. Circ. Res. 103: 1232−1240. 2008. https://doi.org/10.1161/01.RES.0000338597.71702.ad -
Suliman H.B., Carraway M.S., Tatro L.G., Piantadosi C.A. A new activating role for CO in cardiac mitochondrial biogenesis. J. Cell Sci. 120:
299−308. 2007. https://doi.org/10.1242/jcs.03318 -
Dominic E.A., Ramezani A., Anker S.D., Verma M., Mehta N., Rao M. Mitochondrial cytopathies and cardiovascular disease. Heart. 100: 611−618. 2014.
https://doi.org/10.1136/heartjnl-2013-304657 -
Muthusamy V.R., Kannan S., Sadhaasivam K., Gounder S.S., Davidson C.J., Boeheme C.,
Hoidal J.R., Wang L., Rajasekaran N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms
in the myocardium. Free Radic. Biol. Med. 52: 366−376. 2012. https://doi.org/10.1016/j.freeradbiomed.2011.10.440 -
Erkens R., Kramer C.M., Lückstädt W., Panknin C., Krause L., Weidenbach M., Dirzka
J., Krenz T., Mergia E., Suvorava T., Kelm M., Cortese-Krott M.M. Left ventricular diastolic dysfunction in Nrf2 knock out mice is associated with
cardiac hypertrophy, decreased expression of SERCA2a, and preserved endothelial function.
Free Radic. Biol. Med. 89: 906−917. 2015. https://doi.org/10.1016/j.freeradbiomed.2015.10.409 -
Ying Z., Chen M., Xie X., Wang X., Kherada N., Desikan R., Mihai G., Burns P., Sun
Q., Rajagopalan S. Lipoicmethylenedioxyphenol reduces experimental atherosclerosis through activation
of Nrf2 signaling. PLoS One. 11: e0148305. 2016. https://doi.org/10.1371/journal.pone.0148305 -
Dickinson S.E., Lin Y., Dedek M., Morrissy S., Johnson J., Chen Q.M. Induction of antioxidant and detoxification response by oxidants in cardiomyocytes:
evidence from gene expression profiling and activation of Nrf2 transcription factor.
J. Mol. Cell Cardiol. 42: 159−176. 2007. https://doi.org/10.1016/j.yjmcc.2006.09.012 -
Wang W., Li S., Wang H., Li B., Shao L., Lai Y, Horvath G., Wang Q., Yamamoto M.,
Janicki J.S., Wang X.L, Tang D., Cui T. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J. Mol. Cell
Cardiol. 72: 305−315. 2014. https://doi.org/10.1016/j.yjmcc.2014.04.006 -
Коршунова А.Ю. Патогенетические особенности клеточной гибели при альтерации миокарда различного
генеза. Дисс. канд. наук, Москва, 141 с. 2016. [Korshunova A.Yu. Patogeneticheskie osobennosti kletochnoj gibeli pri al`teracii miokarda razlichnogo
geneza. Diss. kand. nauk. Moskva, 141 s. 2016. (In Russ)]. -
Xing Y., Niu T., Wang W., Li J., Li S., Janicki J.S., Ruiz S., Meyer C.J., Wang X.L.,
Tang D., Zhao Y., Cui T. Triterpenoid dihydro-CDDO-trifluoroethyl amide protects against maladaptive cardiac
remodeling and dysfunction in mice: a critical role of Nrf2. PLoS One. 7. 9: 1−8.
2012. https://doi.org/10.1371/journal.pone.0044899 -
Suzuki T., Shibata T., Takaya K., Shiraishi K., Kohno T., Kunitoh H., Tsuta K., Furuta
K., Goto K., Hosoda F., Sakamoto H., Motohashi H., Yamamoto M. Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression
levels. Mol. Cell Biol. 33. 2402−2412. 2013. https://doi.org/10.1128/MCB.00065-13 -
Морозова К.В. Полиморфизм генов ферментов детоксикации, антиоксидантной защиты и репарации ДНК
в генезе невынашивания беременности. Автореф. канд. дисс. Москва, 2014 [Morozova K.V. Polimorfizm genov fermentov detoksikacii, antioksidantnoj zashhity` i reparacii DNK
v geneze nevy`nashivaniya beremennosti. Avtoref. kand. diss. Moskva, 2014. (In Russ)]. -
Demeulder B., Zarrinpashneh E., Ginion A., Viollet B., Hue L., Rider M.H., Vanoverschelde
J.L., Beauloye C., Horman S., Bertrand L. Differential regulation of eEF2 and p70S6K by AMPKalpha2 in heart. Biochim. Biophys.
Acta. 1832: 780−790. 2013. https://doi.org/10.1016/j.bbadis.2013.02.015 -
Sciarretta S., Yee D., Ammann P., Nagarajan N., Volpe M., Frati G., Sadoshima J. Role of NADPH oxidase in the regulation of autophagy in cardiomyocytes. Clin Sci
(Lond). 128: 387−403. 2015. https://doi.org/10.1042/CS20140336 -
Garlid K.D., Dos Santos P., Xie Z.J., Costa A.D., Paucek P. Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim. Biophys. Acta. 1606: 1–21.
2003. https://doi.org/10.1016/S0005-2728(03)00109-9 -
Mitchell P., Moyle J. Translocation of some anions cations and acids in rat liver mitochondria. Eur. J.
Biochem. 9: 149−155. 1969. https://doi.org/10.1111/j.1432-1033.1969.tb00588.x -
Chaves E., Yung D., Brierley G. Energy-dependent exchange of K+ in heart mitochondria, K+ efflux.Arch. Bioch. Biophys. 183: 460−470. 1977. https://doi.org/10.1016/0003-9861(77)90381-2
-
Diwan J.J., Haley T., Sanadi D.R. Reconstitution of K+ transport with 53 kDa mitochondrial protein. Biochem Biophys Res Commun., 153: 224−230.
1988. https://doi.org/10.1016/S0006-291X(88)81212-9 -
Murry C.E., Jennings R.B., Reimer K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium.
Circulation. 74: 1124−1136. 1986. https://doi.org/10.1161/01.CIR.74.5.1124 -
Santillo E., Migale M., Postacchini D., Balestrini F., Incalzi R.A. Cardioprotection by conditioning mimetic drugs. Antiinflamm. Antiallergy Agents Med.
Chem. 15: 15−30. 2016. https://doi.org/10.2174/1871523015666160719155122 -
Лихванцев В.В., Мороз В.В., Гребенчиков О.А., Гороховатский Ю.И., Заржецкий Ю.В., Тимошин С.С., Левиков Д.И., Шайбакова В.Л. Ишемическое и фармакологическое прекондиционирование. Общая реаниматология. 7 (6):
59−65. 2011. [Lixvancev V.V., Moroz V.V., Grebenchikov O.A., Goroxovatskij Yu.I., Zarzheczkij Yu.V.,
Timoshin S.S., Levikov D.I., Shajbakova V.L. Ishemicheskoe i farmakologicheskoe prekondicionirovanie. Obshhaya reanimatologiya.
7 (6): 59−65. 2011. (In Russ)]. https://doi.org/10.15360/1813-9779-2011-6-59 -
Downey J.M., Davis A.M., Cohen M.V. Signaling pathways in ischemic preconditioning. Heart Fail. Rev. 12: 181−188. 2007.
https://doi.org/10.1007/s10741-007-9025-2 -
Xu W., Liu Y., Wang S., McDonald T., Van Eyk J.E., Sidor A., O’Rourke B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science. 298: 1029−1033. 2002.
https://doi.org/10.1126/science.1074360 -
Contreras G.F., Castillo K., Enrique N., Carrasquel-Ursulaez W., Castillo J.P., Milesi
V., Neely A., Alvarez O., Ferreira G., González C., Latorre R. ABK(Slo1)channel journey from molecule to physiology. Channels (Austin.) 7: 442−458.
2013. https://doi.org/10.4161/chan.26242 -
Gao Y.D., Garcia M.L. Interactionofagitoxin2, charybdotoxin, andiberiotoxin with potassium channels: selectivity
between voltage-gated and maxi-K+ channels. Proteins. 52:146−154. 2003. https://doi.org/10.1002/prot.10341 -
Banerjee A., Lee A., Campbell E., MacKinnon R. Structure of apore-blocking toxin in complex with a eukaryotic voltage-dependent
K+ channel. Elife. 2:e00594. 2013. https://doi.org/10.7554/eLife.00594 -
Schreiber M., Salkoff L. A novel calcium-sensing domain in the BK-channel. Biophys. J. 73: 1355−1363. 1997.
https://doi.org/10.1016/S0006-3495(97)78168-2 -
Xia X.M., Zeng X., Lingle C.J. Multiple regulatory sites in large- conductance calcium-activated potassium channels.
Nature. 418: 880−884. 2002. https://doi.org/10.1038/nature00956 -
Yang H., Shi J., Zhang G., Yang J., Delaloye K., Cui J. Activation of Slo1BK-channels by Mg2+ coordinated between the voltage sensorand RCK1 domains. Nat. Struct. Mol. Biol. 15:
1152−1159. 2008. https://doi.org/10.1038/nsmb.1507 -
Balderas E., Zhang J., Stefani E., Toro L. Mitochondrial BKCa channel. Front. Physiol. 6:104. 2015. https://doi.org/10.3389/fphys.2015.00104
-
Siemen D., Loupatatzis C., Borecky J.,Gulbins E., Lang F. Ca2+-activated K+ channel of the BK-type in the inner mitochondrial membrane of a human glioma cell
line. Biochim. Biophys. Res. Comm. 257: 549−554. 1999. https://doi.org/10.1006/bbrc.1999.0496 -
Singh H., Lu R., Bopassa J.C., Meredith A.L., Stefani E., Toro L. mitoBK Ca is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial
location. Proc. Natl. Acad. Sci.U.S.A. 110: 10836−10841. 2013. https://doi.org/10.1073/pnas.1302028110 -
Gu X.Q., Pamenter M.E., Siemen D., Sun X., Haddad G.G. Mitochondrial but not plasmalemmal BK channels are hypoxia-sensitive in human glioma.
Glia. 62: 504−513. 2014. https://doi.org/10.1002/glia.22620 -
Tanaka Y., Meera P., Song M., Knaus H.-G., Toro L. Molecular constituents of maxiKCa channels in human coronary smooth muscle: predominant
alpha + beta subunit complexes. J. Physiol. 502: 545−557. 1997. https://doi.org/10.1111/j.1469-7793.1997.545bj.x -
Schmitt N., Grunnet M., Olesen S.P. Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol.
Rev. 94: 609−653. 2014. https://doi.org/10.1152/physrev.00022.2013 -
Kulawiak B., Kudin A.P., Szewczyk A., Kunz W.S. BK-channel openers inhibit ROS production of isolated rat brain mitochondria. Exp.
Neurol. 212:543−547. 2008. https://doi.org/10.1016/j.expneurol.2008.05.004 -
Cheng Y., Gulbins E., Siemen D. Activation of the permeability transition pore by Bax via inhibition of the mitochondrial
BK-channel. Cell. Physiol. Biochem. 27: 191−200. 2011. https://doi.org/10.1159/000327944 -
Cheng Y., Gu X.Q., Bednarczyk P., Wiedemann F.R., Haddad G.G., Siemen D. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane
and reduces activity of the permeability transition pore. Cell. Physiol. Biochem.
22: 127−136. 2008. https://doi.org/10.1159/000149790
Дополнительные материалы отсутствуют.
- Авторы
- Резюме
- Файлы
- Ключевые слова
- Литература
Харечкина Е.С.
1
Никифорова А.Б.
1
1 ФГБУН «Институт теоретической и экспериментальной биофизики РАН
Настоящая обзорная статья рассматривает существующие в настоящее время представления о механизмах, которые лежат в основе генерации активных форм кислорода при пермеабилизации митохондриальных мембран. Рассмотрена роль ионов кальция и комплексов дыхательной цепи митохондрий. Обсуждается влияние уровня пиридиновых нуклеотидов, компонентов антиоксидантной системы, а также участие матриксных Са2+-активируемых дегидрогеназ. В литературе имеются данные, показывающие, что индукция митохондриальной Са2+-зависимой поры вызывает конформационные перестройки дыхательных комплексов I, II и III, что усиливает генерацию активных форм кислорода. Вход кальция в матрикс митохондрий может увеличивать скорости продукции активных форм кислорода за счет активации пируватдегидрогеназы и а-кетоглутаратдегидрогеназы, а также способствовать выходу цитохрома с в цитозоль при индукции митохондриальной поры. Выход глутатиона и восстановленных пиридиновых нуклеотидов через пору снижает антиоксидантную защиту матрикса митохондрий и увеличивает продукцию супероксид аниона и перекиси водорода. Явление всплеска активных форм кислорода, вызванного пермеабилизацией митохондрий, сопровождает различные патологические состояния, включая ишемию с последующей реперфузией, поэтому понимание молекулярных процессов, лежащих в его основе, необходимо для дальнейшей разработки способов его фармакологической коррекции.
активные формы кислорода
митохондриальная пора
nadh
nadph
дыхательная цепь митохондрий
1. Halestrap A.P., Richardson A.P. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury // Journal of Molecular and Cellular Cardiology. 2015. Vol. 78. P. 129-141.
2. Brookes P.S., Yoon Y., Robotham J.L. et al. Calcium, ATP, and ROS: a mitochondrial love-hate triangle // American Journal of Physiology. Cell Physiology. 2004. Vol. 287 (4). P. 817-833.
3. Ruiz-Ramírez A., López-Acosta O., Barrios-Maya M.A., El-Hafidi M. Cell death and heart failure in obesity: role of uncoupling proteins // Oxidative Medicine and Cellular Longevity. 2016. Vol. 2016. P. 1-11.
4. Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release // Physiological Reviews. 2014. Vol. 94 (4). P. 909-950.
5. Andrienko T., Pasdois P., Rossbach A., Halestrap A.P. Real-time fluorescence measurements of ROS and [Ca2+] in ischemic/reperfused rat hearts: detectable increases occur only after mitochondrial pore opening and are attenuated by ischemic preconditioning // PLoS ONE. 2016. Vol. 11 (12).
6. Korge P., John S.A., Calmettes G., Weiss J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: the role of complex II // The Journal of Biological Chemistry. 2017. Vol. 292 (24). P. 9896-9905.
7. Korge P., Calmettes G., John S.A., Weiss J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: The role of complex III // The Journal of Biological Chemistry. 2017. Vol. 292 (24). P. 9882-9895.
8. Batandier C., Leverve X., Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I // The Journal of Biological Chemistry. 2004. Vol. 279 (17). P. 17197-17294.
9. Cadenas S. ROS and redox signaling in myocardial ischemia reperfusion injury and cardioprotection // Free Radical Biology and Medicine. 2018. Vol. 117. P. 76-89.
10. Chouchani E.T., Pell V.R., James A.M. et al. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury // Cell Metabolism. 2016. Vol. 23 (2). P. 254-263.
11. Гривенникова В.Г., Виноградов А.Д. Генерация активных форм кислорода митохондриями // Успехи биологической химии. 2013. Т. 53. С. 245-296.
12. Maklashina E., Sher Y., Zhou H.Z. et al. Effect of anoxia/reperfusion on the reversible active/de-active transition of NADH-ubiquinone oxidoreductase (complex I) in rat heart // Biochimica et Biophysica Acta. 2002. Vol. 1556 (1). P. 6-12.
13. Grivennikova V.G., Kareyeva A.V., Vinogradov A.D. What are the sources of hydrogen peroxide production by heart mitochondria? // Biochimica et Biophysica Acta. 2010. Vol. 1797 (6-7). P. 939-944.
14. Chouchani E.T., Methner C., Nadtochiy S.M. et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I // Nature Medicine. 2013. Vol. 19 (6). P. 753-759.
15. Imlay, J.A. A metabolic enzyme that rapidly produces superoxide, fumarate reductase of Escherichia coli // Journal of Biological Chemistry. 1995. Vol. 270. P. 19767-19777.
16. Siebels I., Drose S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates // Biochimica et Biophysica Acta. 2013. Vol. 1827 (10). P. 1156-1164.
17. Quinlan C.L., Orr A.L., Perevoshchikova I.V. et al. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions // Journal of Biological Chemistry. 2012. Vol. 287 (32). P. 27255-27264.
18. Grivennikova V.G., Kozlovsky V.S., Vinogradov A.D. Respiratory complex II: ROS production and the kinetics of ubiquinone reduction // Biochimica et Biophysica Acta. 2017. Vol. 1858 (2). P. 109-117.
19. Chouchani E.T., Pell V.R., Gaude E. et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS // Nature. 2014. Vol. 515. P. 431-435.
20. Lemarie A., Huc L., Pazarentzos E. et al. Specific disintegration of complex II succinate:ubiquinone oxidoreductase links pH changes to oxidative stress for apoptosis induction // Cell Death and Differentiation. 2011. Vol. 18 (2). P. 338-349.
21. Huang L.S., Cobessi D., Tung E.Y., Berry E.A. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern // Journal of Molecular Biology. 2005. Vol. 351 (3). P. 573-597.
22. Vercesi A.E. The participation of NADP, the transmembrane potential and the energy-linked NAD(P) transhydrogenase in the process of Ca2+ efflux from rat liver mitochondria // Archives of Biochemistry and Biophysics. 1987. Vol. 252 (1). P. 171-178.
23. Peng T.I., Jou M.J. Oxidative stress caused by mitochondrial calcium overload // Annals of the New York Academy of Sciences. 2010. Vol. 1201. P. 183-188.
24. Starkov A.A. An update on the role of mitochondrial α-ketoglutarate dehydrogenase in oxidative stress // Molecular and Cellular Neuroscience. 2013. Vol. 55. P. 13-16.
25. Nickel A.G., von Hardenberg A., Hohl M. et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure // Cell Metabolism. 2015. Vol. 22 (3). P. 472-484.
26. Wei A.C., Liu T., Winslow R.L., O’Rourke B. Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering // Journal of General Physiology. 2012. Vol. 139 (6). P. 465-478.
27. Denton R.M. Regulation of mitochondrial dehydrogenases by calcium ions // Biochimica et Biophysica Acta. 2009. Vol. 1787 (11). P. 1309-1316.
28. Patterson S.D., Spahr C.S., Daugas E. et al. Mass spectrometric identification of proteins released from mitochondria undergoing permeability transition // Cell Death and Differentiation. 2000. Vol. 7 (2). P. 137–144.
29. Ott M., Robertson J.D., Gogvadze V. et al. Cytochrome c release from mitochondria proceeds by a two-step process // Proceedings of the National Academy of Sciences of the United States of America. 2002. Vol. 99 (3). P. 1259–1263.
30. Pereverzev M.O., Vygodina T.V., Konstantinov A.A., Skulachev V.P. Cytochrome c, an ideal antioxidant // Biochemical Society Transactions. 2003. Vol. 31. Pt. 6. P. 1312–1315.
Пермеабилизацию внешней мембраны митохондрий опрeделяют как резкое увеличение ее проницаемости для ионов и растворов массой менее 1,5 kDa, приводящее к потере мембранного потенциала, набуханию митохондрий, разрыву их внешней мембраны и выходу апоптогенных факторов. Этот процесс происходит после открывания мегаканала, известного как Са2+-зависимая неспецифическая митохондриальная пора (mPTP) [1]. Открывание mPTP, по-видимому, является ключевым фактором, вызывающим клеточную гибель и необратимые повреждения органов при многих патологических состояниях, таких как ишемия с последующей реперфузией, нейродегенеративные заболевания, мышечная дистрофия.
Главным активатором mPTP является кальций, при этом чувствительность к катиону многократно увеличивается при окислительном стрессе [2]. Такие условия наблюдаются при ишемии/реперфузии, и считается, что они являются главным триггером открывания mPTP. Предположение о том, что основной всплеск активных форм кислорода (АФК) происходит при открывании поры и после, долгое время ставилось под сомнение, так как известно, что ее индукция приводит к разобщению митохондрий, а это, в свою очередь, снижает продукцию АФК [3]. Однако группой Д. Зорова было обнаружено, что аккумулирование АФК в матриксе митохондрий сердечных миоцитов при фотоактивации тетраметилродаминовых производных запускает индукцию mPTP, которая сопровождается многократно усиленной продукцией («всплеском») АФК. Данное явление авторы назвали АФК-индуцированный выход АФК («ROS — induced ROS releas» (RIRR)) [4]. Впоследствии появилось много работ, демонстрирующих всплеск АФК, вызванный индукцией mPTP [5-8]. Выход АФК в цитозоль может активировать редокс-чувствительные ферменты, а также запускать сложный сигнальный ответ и генерацию АФК в соседних митохондриях. Данный процесс имеет важное физиологическое и патологическое значение, поскольку может индуцировать гибель не только старых и поврежденных митохондрий и клеток, но и здоровых. Вопрос о путях образования АФК при индукции mPTP несет важную научную и практическую значимость, но к настоящему моменту остается открытым.
Цель исследования
Произвести обзор существующих в современной литературе данных и гипотез о сайтах и механизмах продукции АФК при пермеабилизации внешней мембраны митохондрий.
Комплекс I дыхательной цепи митохондрий
Комплекс I (НАДН-убихинон оксидоредуктаза) является одним из главных мест продукции АФК в митохондриях. Считается, что основными сайтами генерации АФК в нем выступают флавинмононуклеотид НАДН-связывающего сайта (сайт If), и убисемихинон коэнзим Q-связывающего сайта (сайт Iq) [9]. Продукция супероксида на сайте If происходит во время прямого транспорта электронов, когда ФМН находится в сильно восстановленном состоянии и зависит от соотношения НАДН/НАД+ в матриксе. Ингибитор коэнзим Q-связывающего сайта ротенон увеличивает продукцию супероксида, так как вызывает возвращение электронов на ФМН. Продукция супероксида на комплексе I также происходит во время обратного транспорта электронов, когда пул коэнзима Q полностью восстановлен [10].
При патологических условиях увеличение эффективности АФК-генерирующих сайтов комплекса I могут быть связаны с его конформационными перестройками. Открывание mPTP сильно снижает ротенон-чувствительную активность НАДН-убихинон редуктазы и увеличивает продукцию Н2О2 в присутствии ≥50 µМ НАДН [8]. НАДН-убихинон оксидоредуктаза характеризуется медленным переходом из активного состояния в неактивное и наоборот. Это предполагает большие конформационные перестройки комплекса, по крайней мере той его части, которая вовлечена в ротенон-чувствительное восстановление убихинона [11]. Было показано, что комплекс I, выделенный из сердца крыс, подвергшегося 30-минутной аноксичной перфузии, переходил в неактивное состояние и возвращался к активному после реоксигенации [12]. Авторы предположили, что эти конформационные перестройки могут быть связаны с генерацией АФК после того, как ткани сердца, подвергшиеся коронарной окклюзии, реоксигенируются. Переход комплекса в неактивное состояние сопровождается специфическим демаскированием Cys39 субъединицы ND3 [13]. Было показано, что нитрозирующие соединения, обратимо модифицирующие данный цистеин, могут использоваться в качестве фармакологической защиты от генерации АФК при реперфузии [14].
Комплекс II дыхательной цепи митохондрий
Комплекс II, или сукцинат-убихинон оксидоредуктаза, является тетрамерным, содержащим железо-серные кластеры флавопротеином внутренней мембраны митохондрий. Он одновременно участвует в работе цикла Кребса и дыхательной цепи, осуществляя превращение сукцината в фумарат и восстанавливая убихинон до убихинола.
Возможность образования АФК флавином фумаратредуктазы E. coli (сайт IIf) в присутствии низких концентраций дикарбоновых кислот впервые была показана в работе [15]. Впоследствии продукция АФК была продемонстрирована на субмитохондриальных частицах митохондрий бычьего сердца [16] и скелетных мышц [17]. Ингибитор комплекса II атпенин А5 и ингибитор комплекса III стигмателлин, который блокирует окисление убихинола комплексом III, стимулируют продукцию АФК комплексом II в присутствии сукцината. Малонат, напротив, ингибирует генерацию АФК комплексом II, что указывает на то, что АФК образуются на полностью восстановленном флавиновом сайте IIf [16], хотя не исключены и другие сайты [18]. Зависимость продукции перекиси водорода от концентрации сукцината имеет колоколообразную форму: уровень перекиси растет с увеличением концентрации субстрата до 400 μМ, затем значительно снижается при миллимолярных концентрациях, обычно используемых для энергизации митохондрий. Причиной этого явления является то, что комплекс II генерирует АФК только тогда, когда его флавиновый сайт IIf не занят дикарбоновыми кислотами [16]. Cукцинат и другие интермедиаты цикла Кребса, которые взаимодействуют с сайтом связывания дикарбоновых кислот, могут ограничивать доступ к нему кислорода и, таким образом, подавлять продукцию АФК комплексом II. Уровень сукцината и фумарата в матриксе увеличивается во время ишемии/гипоксии, однако это не предотвращает образование АФК. Напротив, было показано, что аккумулирование сукцината во время ишемии сильно коррелирует с продукцией АФК и повреждениями при реперфузии [19]. Авторы предположили, что главным источником АФК в данных условиях является обратный поток электронов через комплекс I [10]. Однако, в условиях длительной ишемии, когда мембраны полностью деполяризуются, данный механизм вряд ли осуществим. Альтернативный механизм генерации АФК предполагает получение доступа кислорода к восстановленному сайту IIf из-за снижения содержания дикарбоновых кислот в его непосредственной близости в результате ускорения выхода сукцината и фумарата из матрикса при индукции mPTP [6]. Данный механизм требует ингибирования комплекса II на уровне восстановления убихинона либо ингибирования окисления убихинола комплексом III.
Конформационные перестройки комплекса II также могут способствовать всплеску АФК при пермеабилизации мембран. Было показано, что при понижении внутриклеточного рН, наблюдающегося при апоптозе, происходит диссоциация комплекса II: субъединицы сукцинатдегидрогеназы SDHA и SDHB, осуществляющие окисление сукцината до фумарата и перенос электронов через железо-серные кластеры, отделяются от сайта восстановления коэнзима Q сукцинат CoQ оксидоредуктазы (SQR) [20]. Это приводит к ингибированию активности SQR, при этом сукцинатдегидрогеназная активность остается в норме. Такая диссоциация приводит к прямому одноэлектронному восстановлению кислорода железо-серным кластером комплекса II. И хотя известно, что низкий рН является ингибитором mPTP, тем не менее данный механизм всплеска АФК может иметь место при ишемии, когда происходит падение рН. В это время могут происходить конформационные перестройки комплекса II, и впоследствии, при реперфузии, когда рН восстанавливается до исходного уровня, открывается mPTP и наблюдается всплеск АФК, образуемых на диссоциированном комплексе.
Комплекс III дыхательной цепи митохондрий
Комплекс III (убихинол-цитохром с оксидоредуктаза) – еще один возможный сайт образования АФК. Данный белок осуществляет перенос электронов от убихинона на цитохром с в процессе функционирования так называемого Q-цикла. В ходе данного процесса происходит образование нестабильного семихинона, который может передавать электрон на кислород, образуя при этом супероксидный радикал. Однако в нормальных условиях такая реакция маловероятна, так как семихинон быстро окисляется цитохромом b. Резкое возрастание уровня супероксида происходит при ингибировании комплекса антимицином А, а также при ишемии длительностью более 30 минут [7]. Одной из причин данного явления могут быть его конформационные перестройки, вызванные связыванием ингибитора [21]. На изолированных митохондриях сердца было показано, что комплекс III, заингибированный с помощью антимицина A, генерирует значительное количество АФК в присутствии Mg2+ и НАД+ и в отсутствии экзогенных субстратов при индукции mPTP кальцием и аламетицином. Авторы показали, что в этих условиях продукция перекиси водорода относится к Mg2+-зависимой генерации НАДН малатдегидрогеназой. Продукция H2O2 ингибировалась стигмателлином и пирицидином, что указывает на важность НАДН-зависимого восстановления убихинона для генерации АФК в данных условиях. Эти данные подтверждают гипотезу, согласно которой во время ишемии при индукции mPTP увеличение концентрации Mg2+, НАД+ в матриксе активирует малатдегидрогеназу, которая восстанавливает НАД+, используя малат, концентрация которого повышается вследствие увеличения уровня сукцината и фумарата. Восстановленные эквиваленты поступают на заингибированный комплекс III, в результате чего происходит всплеск АФК [7].
Роль пиридиновых нуклеотидов в генерации АФК
Раннее было показано, что окисление НАД(Ф)Н матрикса митохондрий предшествует открыванию mPTP [22]. Кроме того, индукция поры приводит к утечке пиридиновых нуклеотидов в цитозоль клетки [23]. Данное изменение баланса НАД(Ф)Н должно влиять на продукцию АФК при пермеабилизации митохондрий. Зависимость генерации АФК от концентрации НАДН была исследована группой А. Виноградова. Было показано, что максимальная продукция супероксида достигает максимума при концентрации НАДН 10-50 μМ, при миллимолярных концентрациях продукция радикала тормозится [11]. Так как физиологические концентрации НАДН/НАД+ пары матрикса находятся в миллимолярном диапазоне, то вклад комплекса I в генерацию АФК в нормальных условиях может быть незначительным. Было обнаружено, что в пермеабилизованных митохондриях происходит высокая, зависящая от отношения НАД(Ф)Н/НАД(Ф)+ и стимулируемая ионами аммония продукция Н2О2. При этом выход перекиси водорода был нечувствителен к дикумаролу (ингибитору НАДН-хинон оксидоредуктазы) и НАДН-OH (ингибитору комплекса I), что указывает на матриксную локализацию H2O2-генерирующего сайта. Исследуемый белок обладал НАДН:липоамид оксидоредуктазной активностью и был идентифицирован как дигидролипоамиддегидрогеназа [11]. Данный белок является важным компонентом (так называемым Е3 компонентом) двух ФАД-cодержащих митохондриальных ферментов: а-кетоглутаратдегидрогеназного комплекса и пируватдегидрогеназного комплекса. Согласно данным, полученным на очищенных комплексах и на изолированных митохондриях [24], компонент Е3 отвечает за продукцию супероксида и перекиси водорода. Было показано, что пермеабилизованные митохондрии сердца крыс, окисляющие НАДН, продуцируют около 50% перекиси водорода за счет работы комплекса I, а остальные 50% приходятся на долю дигидролипоамиддегидрогеназы [13].
Восстановленные формы пиридиновых нуклеотидов не только поставляют электроны в дыхательную цепь митохондрий, но также регулируют редокс-статус матрикса через про- и антиоксидантные белки. Одним из таких белков является глутатион, который, совместно с НАДФН, является субстратом антиоксидантных белков глутатионпероксидазы и глутатионредуктазы [2]. При открывании mPTP может происходит выход НАДФH и глутатиона, что вызывает накопление Н2О2 [23]. Более того, в данных условиях из-за падения мембранного потенциала никотинамиднуклеотидтрансгидрогеназа (НАДФН- трансгидрогеназа) не может поддерживать высокий уровень восстановленного НАДФ+, что способствует окислительного стрессу [22]. В физиологических условиях данный фермент осуществляет регенерацию НАДФН в прямой реакции, используя НАДН в качестве субстрата. Эта реакция энергетически выгодна, поскольку трансгидрогенирование между НАДН и НАДФН связано с протонным градиентом вдоль внутренней мембраны. Однако в патологических условиях она может протекать в обратном направлении, регенерируя НАДН для синтеза ATP за счет утилизации НАДФН [25]. Таким образом, антиоксидантная защита, связанная с уровнем восстановленности НАДФ+, падает, что способствует продукции H2O2.
Роль кальция в генерации АФК
Известно, что увеличение концентрации кальция в матриксе митохондрий запускает индукцию mPTP, при этом чувствительность поры к катиону увеличивается при окислительном стрессе, повышением уровня фосфата и снижением пула адениновых нуклеотидов [1]. Концентрация ионов кальция в матриксе митохондрий находится в пределах примерно 10 nМ. При этом их кальциевая емкость очень высока, изолированные митохондрии способны секвестрировать более 1M кальция из среды, поддерживая концентрацию свободного кальция в микромолярных пределах, в которых происходит регуляция Ca2+-зависимых ферментов [26]. К таким ферментам относятся пируватдегидрогеназа и а-кетоглутаратдегидрогеназа. Их активация приводит к усилению дыхания и синтеза АТФ и, вероятно, к повышению продукции АФК [24; 27].
В процессе пермеабилизации митохондриальных мембран происходит выход из межмембранного пространства и матрикса примерно 100 белков, в том числе таких важных элементов антиоксидантной защиты, как глутатион и цитохром с [28].
Цитохром с является положительно заряженным белком, который связан с кардиолипином на внешней стороне внутренней мембраны митохондрий, а также с дыхательными комплексами III и IV. Было показано, что выход цитохрома с является двухступенчатым процессом, включающим отсоединение белка от внутримембранных связывающих сайтов и последующую его транслокацию через внешнюю мембрану [29]. Ca2+ может усиливать диссоциацию цитохрома с от внутренней мембраны, так как является его конкурентом за связывание с отрицательно заряженным кардиолипином. Это способствует выходу цитохрома с в цитозоль при индукции mPTP. Более того, АФК, образуемые при пермеабилизации мембран, могут вызывать окисление кардиолипина, приводящее к изменению его физических свойств, что также может усиливать выход цитохрома с из митохондрий и способствовать еще большей генерации АФК. Пониженный уровень белка замедляет транспорт электронов от комплекса III к комплексу IV и, таким образом, увеличивает продукцию АФК в Q-цикле. Кроме того, цитохром с сам по себе является эффективным антиоксидантом, способным эффективно восстанавливаться супероксид анионом [30]. Таким образом, повышение концентрации кальция в митохондриях оказывает стимулирующее влияние на АФК-продуцирующие ферменты матрикса и приводит к падению антиоксидантной защиты, тем самым увеличивая общий уровень АФК, генерируемый митохондриями.
Заключение
Митохондрии являются одновременно потенциальным источником и мишенью действия АФК, приводящим к потере митохондриальных функций и, как следствие, к необратимому повреждению клеток при многих патологических процессах. Важную роль при этом играет mPTP, индукция которой может приводить к мощной генерации АФК, оказывающих повреждающее действие на соседние органеллы и целые клетки. В настоящее время причины данного явления слабо изучены, хотя в литературе имеется несколько гипотез. Предполагается, что в основе всплеска АФК могут лежать конформационные перестройки комплексов дыхательной цепи, активация дегидрогеназ матрикса в результате действия Са2+, изменение баланса НАД(Ф)Н/НАД(Ф)+ матрикса и истощение антиоксидантной системы. Дальнейшее исследование механизмов и сайтов продукции АФК при индукции mPTP представляется необходимым, поскольку их точное определение позволит разработать способы их регуляции для предупреждения развития многих патологических состояний организма.
Работа выполнена при поддержке гранта РНФ № 17-75-10122.
Библиографическая ссылка
Харечкина Е.С., Никифорова А.Б. МЕХАНИЗМЫ ГЕНЕРАЦИИ АКТИВНЫХ ФОРМ КИСЛОРОДА ПРИ ПЕРМЕАБИЛИЗАЦИИ МИТОХОНДРИАЛЬНЫХ МЕМБРАН // Современные проблемы науки и образования. – 2018. – № 4.
;
URL: https://science-education.ru/ru/article/view?id=27719 (дата обращения: 22.03.2023).
Предлагаем вашему вниманию журналы, издающиеся в издательстве «Академия Естествознания»
(Высокий импакт-фактор РИНЦ, тематика журналов охватывает все научные направления)





